Articles
- Page Path
- HOME > Endocrinol Metab > Volume 38(6); 2023 > Article
-
Review ArticleMiscellaneous Toward Systems-Level Metabolic Analysis in Endocrine Disorders and Cancer
 Keypoint
Keypoint
This review explores the interplay between the endocrine system and systems-level metabolism in health and disease. The authors highlight metabolic fluxes in conditions like obesity, diabetes, Cushing's syndrome, and cancers. Recent advances in metabolomics, fluxomics, and systems biology have the potential for new insights into dynamic metabolism, offering potential biomarkers, therapeutic targets, and personalized medicine. -
Aliya Lakhani1
 , Da Hyun Kang2, Yea Eun Kang2, Junyoung O. Park1
, Da Hyun Kang2, Yea Eun Kang2, Junyoung O. Park1 -
Endocrinology and Metabolism 2023;38(6):619-630.
DOI: https://doi.org/10.3803/EnM.2023.1814
Published online: November 21, 2023
1Department of Chemical and Biomolecular Engineering, University of California, Los Angeles, Los Angeles, CA, USA
2Department of Internal Medicine, Chungnam National University College of Medicine, Daejeon, Korea
- Corresponding authors: Yea Eun Kang. Department of Internal Medicine, Chungnam National University College of Medicine, 282 Munhwa-ro, Jung-gu, Daejeon 35015, Korea Tel: +82-42-2807148, Fax: +82-42-280-7996, E-mail: yeeuni2200@gmail.com
- Junyoung O. Park. Department of Chemical and Biomolecular Engineering, University of California, Los Angeles, Los Angeles, CA 90095, USA Tel: +1-310-267-5746, Fax: +1-310-206-4107, E-mail: jop@ucla.edu
• Received: September 9, 2023 • Revised: October 27, 2023 • Accepted: November 1, 2023
Copyright © 2023 Korean Endocrine Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
- Metabolism is a dynamic network of biochemical reactions that support systemic homeostasis amidst changing nutritional, environmental, and physical activity factors. The circulatory system facilitates metabolite exchange among organs, while the endocrine system finely tunes metabolism through hormone release. Endocrine disorders like obesity, diabetes, and Cushing’s syndrome disrupt this balance, contributing to systemic inflammation and global health burdens. They accompany metabolic changes on multiple levels from molecular interactions to individual organs to the whole body. Understanding how metabolic fluxes relate to endocrine disorders illuminates the underlying dysregulation. Cancer is increasingly considered a systemic disorder because it not only affects cells in localized tumors but also the whole body, especially in metastasis. In tumorigenesis, cancer-specific mutations and nutrient availability in the tumor microenvironment reprogram cellular metabolism to meet increased energy and biosynthesis needs. Cancer cachexia results in metabolic changes to other organs like muscle, adipose tissue, and liver. This review explores the interplay between the endocrine system and systems-level metabolism in health and disease. We highlight metabolic fluxes in conditions like obesity, diabetes, Cushing’s syndrome, and cancers. Recent advances in metabolomics, fluxomics, and systems biology promise new insights into dynamic metabolism, offering potential biomarkers, therapeutic targets, and personalized medicine.
- Metabolism plays a pivotal role in supporting systemic homeostasis in humans. It adapts to various factors like nutritional levels, surrounding temperatures, and physical exercise. A conduit for metabolism is the circulatory system, which enables transfer of metabolites between organs, resulting in dynamic shifts in the inter-organ metabolic cycle [1,2]. Meanwhile, the endocrine system acts as a master conductor, releasing hormones like insulin, thyroid hormones, sex hormones, and cortisol, to intricately regulate the metabolism of the whole body.
- Endocrine disorders, characterized by abnormal hormone secretion or action, encompass a spectrum of conditions such as diabetes, thyroid dysfunction, adrenal disorders, obesity, and metabolic syndrome, which collectively contribute to systemic inflammation and a global disease burden. These diseases not only impact individual organ systems but also trigger a cascade of molecular events that reverberate throughout the entire organism, resulting in systemic disturbances. Understanding the relationship between metabolism and endocrine disorders has emerged as a promising avenue for deciphering the underlying metabolic dysregulation in organ-organ communications.
- Cancer is increasingly considered as a systemic disorder. Cancer metastasis is a complex process that involves manifold biological interactions and requires the metabolic reprogramming of cancer cells. Cancer cells reprogram metabolic pathway utilization in order to meet increased bioenergetic and biosynthetic demands and to resist oxidative stress for growth and survival [3]. Cancer driver mutations coupled with microenvironmental nutrient availability control metabolic pathways [3,4]. Moreover, metabolites, when aberrantly accumulated, can promote tumor progression and metastasis (e.g., 2-hydroxyglutarate [2HG], fumarate, succinate) [5]. The development of innovative technologies over the last few decades has revealed the heterogeneity and plasticity of tumors and allowed us to uncover metabolic pathways involved in supporting tumor growth.
- The metabolism research community has witnessed remarkable advancements in quantitative and high-throughput measurement tools to gain systems-level insights into various physiological and pathological processes. Metabolic fluxes, defined as the rates at which metabolites flow through metabolic pathways, quantitatively describe cellular functions, energy homeostasis, and overall physiological balance. In endocrine diseases, dysregulation of metabolic fluxes such as disrupted feedback mechanisms and impaired metabolic pathways go hand in hand with altered hormone signaling and pathogenesis. These aberrant metabolic profiles may serve as biomarkers and point to potential therapeutic targets.
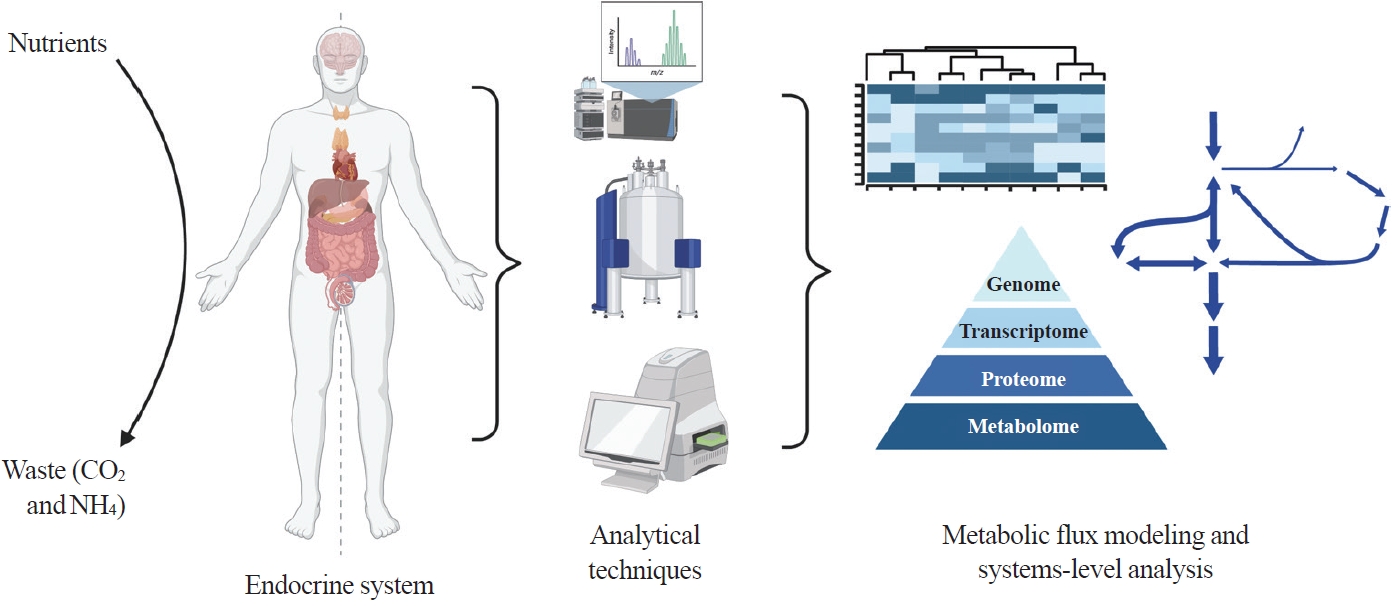
- Here we highlight investigations into the interplay between the endocrine system and systems-level metabolism. We delve into obesity, diabetes, Cushing’s syndrome, and cancers to illustrate metabolic reprogramming in disease initiation, progression, and therapeutic response. Recent advances in metabolomics, fluxomics, and systems biology promise quantitative investigations into such systemic disorders. Systems-level understanding of metabolism in health and disease will unlock novel biomarkers, therapeutic targets, and personalized medicine (Fig. 1).
INTRODUCTION
- Metabolism supports the growth and development of all organisms and requires coordinated regulation of many pathways. In mammals, metabolism contributes to systemic homeostasis. The advent of integrative health has brought our attention to holistic understanding of the systems of the human body and made a profound impact on how we approach diets and medicine for overall health and well-being. Obesity is a type of systemic inflammation accompanied by altered metabolism. Several studies have conducted flux analysis using several types of diets. One such focused on fructose diets in a rat model, which is thought to influence the pathologic condition of obesity [6]. However, organ-organ interactions within obesity are not fully understood because of the complex nature of obesity-related pathophysiology. Recently, metabolomic, proteomic, and transcriptomic data have been used to predict metabolic fluxes; evaluating the livers of fasting leptin-deficient (ob/ob) mice revealed increased gluconeogenic flux in obesity [7].
- Arteriovenous metabolite gradient measurements have been used in obesity to study organ-specific fluxes of metabolites. One study using stable isotope tracing revealed that weight loss from calorie restriction increased glycerol and nonesterified fatty acids released by adipose tissue [8]. Whole-body metabolism of branched-chain amino acids (BCAAs) has also been studied in the context of insulin resistance using high-fat diet-induced (HFD) and leptin receptor-deficient (db/db) obese mice as models. Isotope tracing by liquid chromatography-mass spectrometry (LC-MS) upon 13C valine infusion in db/db mice and 13C isoleucine infusion in HFD mice revealed a reduced BCAA oxidation state in liver and adipose tissues with a shift towards muscle in insulin-resistant mice [9].
- Insulin is an essential hormone that regulates the metabolism of carbohydrates, lipids, and proteins through canonical insulin signaling. A prominent example of systemic disorder is diabetes which impairs systemic glucose homeostasis. Type 1 diabetes mellitus (T1DM) is a chronic immune disorder triggered by the destruction of insulin-producing pancreatic beta cells [10]. Type 2 diabetes mellitus (T2DM) involves insulin resistance, impaired signal transduction in response to insulin stimulation, and impaired insulin secretion.
- Glycolysis and gluconeogenesis are functionally opposite metabolic pathways that together support systemic glucose homeostasis. In T1DM, the absence of insulin leads to uncontrolled gluconeogenesis and excessive glucose production by the liver. In T2DM, insulin resistance in peripheral tissues disrupts glycolytic flux, impairing glucose uptake and utilization. Additionally, dysregulated gluconeogenesis persists even in the presence of hyperglycemia, further exacerbating hyperglycemic states [11]. BCAA metabolism has also played a role with increased levels exacerbating the risk of T1DM development and progression [12,13]. HFDs and high levels of valine, leucine, and isoleucine are associated with insulin resistance in T1DM [14]. T2DM is characterized by increased levels of BCAAs, aromatic amino acids, and glutamate-to-glutamine ratio [15]. To link BCAA catabolism to diabetes, 3-hydroxyisobutyrate has been shown to regulate the trans-endothelial flux of fatty acids [16]. A recent study in mice, however, showed that BCAA catabolism does not affect insulin resistance even though it lowers fasting plasma BCAA levels [17]. Intervention studies based on metabolic fluxes are warranted to clarify mechanisms and explore the extent to which each of these factors leads to pathologic conditions of diabetes.
- To gain mechanistic insights, mathematical models of insulin action and diabetes progression have been developed. The glucose-insulin model (GIM) integrates a system of 1,117 ordinary differential equations and several thousand parameters to model the first steps of glycolytic pathways, allowing for cell proliferation estimates in humans using labeled glucose [18,19]. Beyond glycolysis, whole-body organ-resolved metabolic reconstruction has recently been combined with GIM to analyze metabolic dysregulation with constraints tied to diet, gene expression, and other physiological parameters [20]. This model provides mechanistic insights associated with changes in metabolic fluxes due to exogenous insulin [21]. Other models in conjunction with constraint-based analysis have been used to predict metabolic flux profiles, showing reduced mitochondrial fatty acid oxidation and oxidative phosphorylation in the pancreatic beta cells of diabetic patients [22].
- In T2DM, a study using a rat model identified changes in the hepatic metabolic profile. Proton (1H) nuclear magnetic resonance spectroscopy (NMR) showed rewiring of choline-betainemethionine and amino acid metabolism as well as a decrease in glycolytic activity [23]. In humans, 13C magnetic resonance imaging was used for noninvasive measurement of cardiac pyruvate dehydrogenase fluxes in T2DM patients [24]. Stable isotope tracing with (U-13C6) glucose and gas chromatography-mass spectrometry has been performed to map metabolism in insulin-secreting beta cells. They found that the pentose phosphate pathway (PPP) was upregulated using glucose-6-phosphate (G6P) derived from glycogen [25]. 13C-based metabolic flux analysis (MFA) was used to probe the role of glucose-6-phosphatase catalytic subunit 2 (G6PC2), which converts G6P to glucose. In pancreatic beta cells, G6PC2 was shown to modulate glycolysis and the citric acid cycle, posing as a therapeutic target for enhancing insulin secretion [26]. Another study probed gluconeogenic flux due to dysregulated glucagon signaling found in T2DM. Glucagon was shown to stimulate mitochondrial anaplerotic flux from glutamine, identifying hepatic glutaminase as a new target to treat hyperglycemia [27]. In vivo studies in mice using (U-13C6) fructose showed that high systemic glycerate levels cause glucose intolerance and damage pancreatic beta cells [28]. Maternal/fetal metabolic relationship probed in a recent study revealed that fetal nutrient sourcing was dependent on maternal glycemic state. LCMS and in vivo 13C-glucose tracing showed sorbitol accumulation in fetal tissues and decreased neurotransmitter levels in the fetal brain isolated from dams with maternal hyperglycemia [29].
- Cushing’s syndrome is a rare endocrine disorder that arises from chronic exposure to excessive glucocorticoids [30]. It may be caused by adrenal adenomas, pituitary adenomas, and ectopic sources of adrenocorticotropic hormone (ACTH) production. The hormonal dysregulation in Cushing’s syndrome affects endocrine pathways and disrupts systemic metabolism [31]. Glucocorticoids, primarily cortisol in humans, influence glucose metabolism by promoting gluconeogenesis in the liver, increasing glycogenesis, and modulating insulin sensitivity [32]. Excess glucocorticoid levels disrupt these regulatory mechanisms, leading to increased gluconeogenesis, insulin resistance, and impaired glucose tolerance. Steroid hormone-related metabolites have been identified as early biomarkers for different subtypes of the disease helping to predict progression of Cushing’s syndrome [33,34]; however, metabolic alterations have yet to be fully elucidated. In situ metabolomics of cortisol-producing adrenal adenomas identified the correlation of cortisol and serotonin and tumor size with abundance of fatty acids [35]. 1H-NMR-based metabolomic studies on pituitary adenomas from patients with Cushing’s syndrome revealed metabolic heterogeneity when compared to gonadotropic pituitary adenomas. LC-MS has been used to subtype adrenal or pituitary Cushing’s syndrome by measuring dehydroepiandrosterone sulfate levels, especially when falsely elevated ACTH is suspected [36]. Cushing’s syndrome patients had lower levels of scyllo-inositol, glycine, and phosphoethanolamine and higher levels of aspartate [37]. Metabolic markers can help differentiate pituitary adenomas of various subtypes for noninvasive diagnosis of Cushing’s syndrome.
- Systems-level metabolic alterations in obesity, diabetes, and other endocrine diseases are yet to be fully understood. The incomplete knowledge is due to the complex nature of multi-organ systems, hormone-associated pathophysiology, and the heterogeneity of the effects across humans. To overcome this shortfall, comprehensive and quantitative measurements of metabolic fluxes using integrative omics tools in humans and model systems are needed.
INTEGRATIVE HEALTH AND SYSTEMIC DISORDERS
- Cancer cells exhibit metabolic reprogramming that supports their heightened energetic and biosynthetic demands. Thyroid cancer represents one of the most common endocrine malignancies, characterized by abnormal growth and proliferation of thyroid follicular cells [38]. Over the past decades, substantial progress has been made in elucidating the metabolic alterations that underlie the development and progression of thyroid cancer [39-41].
- Metabolic insights in thyroid cancer hold clinical implications for diagnosis, prognosis, and therapeutic interventions. Inhibiting glycolysis, fatty acid metabolism, or key metabolic enzymes involved in nutrient utilization have emerged as potential strategies to selectively target thyroid cancer cells [41]. Studies have shown that patients with both diabetes and thyroid cancer have smaller tumor sizes when treated with metformin, a drug used for T2DM [42,43]. A recent study investigated metabolic reprogramming in papillary thyroid cancer in vitro and in vivo when treated with metformin. Using an extracellular flux analyzer, metformin was shown to decrease mitochondrial respiration, respiration capacity, aerobic glycolysis, glycolytic capacity, and adenosine triphosphate production [44]. Metabolomics has also elucidated the mechanisms underlying metabolic alterations in different subtypes of thyroid cancer. For example, kinase inhibitors dasatinib and trametinib were shown to alter metabolic flux in anaplastic and papillary thyroid cancer cells [45]. In anaplastic thyroid cells in mice, targeting glycogen metabolism by inhibiting glycogen phosphorylase resulted in increased glucose flux, impairment of PPP, reactive oxygen species accumulation, and eventual cytotoxicity [46].
- One of the most notable metabolic alterations observed in thyroid cancer is increased glycolytic activity, commonly known as the Warburg effect. Overexpression of glycolytic enzymes at the rate-determining steps (i.e., glucose import and phosphorylation, phosphofructokinase, and lactate export) increases glycolytic flux [47]. Thyroid cancer overexpresses hexokinase and has mitochondrial defects, which contribute to heightened glycolysis [48,49]. These defects have been studied in oncocytic thyroid cancer, which is characterized by the accumulation of abnormal mitochondria in the cytoplasm. Recently, a study comparing oncocytic and non-oncocytic thyroid cancer, showed higher glucose uptake and rerouting of glutamine to glutathione for redox homeostasis in the oncocytic cell line to compensate for its defective mitochondrial function [50]. 1H-NMR spectroscopy has been used to show that oxidative stress alters the metabolic profile in thyroid cancer, with increases in lactate and aromatic amino acids and a decrease in citrate [51].
- A recent study elucidated the role of one-carbon metabolism and the serine/glycine metabolic pathway in undifferentiated thyroid cancer using metabolomics, bulk transcriptomics, and singlecell RNA sequencing of cells derived from human tissues [52]. High expression of mitochondrial serine hydroxymethyltransferase (SHMT2) is associated with low thyroid differentiation scores and poor clinical features in thyroid cancer. Inhibition of SHMT2 reduced tumorigenesis in rodent models. Another study reported that genetic inhibition of SHMT2 led to significant inhibition of cell proliferation by depletion of the purine pool [53]. Quantitation of systems-level metabolic fluxes remains an open challenge for the thyroid cancer research community.
- Pheochromocytoma and paraganglioma are rare neuroendocrine tumors that are characterized by excessive catecholamine production [54]. Genetics research has led to discoveries of metabolic alterations associated with these cancers, particularly from mutations in genes in the hypoxia signaling pathways including hypoxia-inducible factor 2α, prolyl hydroxylase domain-containing protein 1/2, succinate dehydrogenase, fumarase, or malate dehydrogenase 2 [55]. A mutation in these genes results in accumulation of succinate, fumarate, and/or 2HG leading to tumorigenesis [56]. New technologies have been used to quantify metabolic changes and discover biomarkers; succinate accumulation, in particular, has been a useful marker in characterizing both tumor type and therapeutic response [57-59]. NMR was used to uncover that high succinate-low glutamate tumors also had high levels of methionine [60]. Furthermore, targeted metabolomics by LC-MS was used to quantify lipid and amino acid metabolic changes in catecholamine-producing tumors from human blood plasma [61]. Recently, serum metabolome profiling of adrenal steroids based on LC-MS and machine learning algorithms has suggested a promising one-step diagnostic approach for the classification of adrenal tumor subtypes [62]. Further study of metabolic pathways involved in adrenal tumors may open up new therapeutic strategies.
- Tumors arise from mutations within proto-oncogenes and tumor suppressor genes. These genetic mutations directly regulate the expression and activity of metabolic enzymes, and the abnormal metabolism of cancer cells also directly affects tumor signal transduction pathways and cellular reactions. Based on this concept, the next-generation anticancer therapeutics examined in many studies and clinical trials target cancer-specific or mutation-specific metabolic phenotypes. Tumors carrying somatic mutations in BRAF and RET genes (constituting the BRAF-like subtype) exhibit a marked extracellular signal-regulated kinase (ERK)-induced transcriptional signature, and tumors carrying either mutations in RAS genes or Pax-8–peroxisome-proliferatoractivated receptor-γ (PPAR-γ) fusion protein (PPFP) display a more pronounced induction of phosphoinositide 3-kinase (PI3K) pathway [63]. In the last decade, alteration of distinct oncogenes has been associated with different tumor cell capacity to undergo metabolic reprogramming, which has emerged as a strong driving force that impacts drug responsiveness and tumor aggressiveness.
- RAS mutation promotes the Warburg effect and activates anabolic pathways [64,65]. Increased efforts have gone into elucidating the molecular underpinnings of altered metabolism in cancer. Oncogenic KRAS promotes glycolytic flux by upregulating several key glycolytic enzymes such as hexokinase 1 and 2, enolase 1 (ENO1), and lactate dehydrogenase-A (LDHA) [66- 68]. KRAS also induces the hexosamine biosynthesis pathway that provides precursors for lipid and protein biosynthesis and the nonoxidative PPP to support increased nucleic acid biosynthesis [69,70]. RAS-driven cancer cells also alter glutaminolysis and rewire the tricarboxylic acid (TCA) cycle, which generates cellular energy and regulates stress granule (SG) formation. KRAS-mutant cancers upregulate glutaminase and NF-E2-related factor 2 (NRF2)-regulated antioxidant genes to drive SG formation, an indicator of chemoresistance [70,71]. This connection of the KRAS-NRF2 axis and glutaminolysis suggests a potential approach to counteract mutant KRAS-mediated drug resistance. Given that the process of SG accumulation is incompletely understood, elucidating the precise underlying mechanisms and metabolic links in RAS-driven cancer will determine the feasibility of SG-based therapeutic strategies.
- A common mutation found in thyroid cancer is BRAFV600E [72-74]. BRAFV600E mutations occur in multiple cancers, such as lung cancer, renal cancer, colorectal cancer, and multiple myeloma. Overexpression of BRAFV600E showed decreased mitochondrial respiration but increased aerobic glycolysis [75]. Data supporting the use of BRAF/mitogen-activated extracellular signal-regulated kinase (MEK) inhibitors that target BRAFV600E mutations in the mitogen-activated protein kinase pathway have begun to emerge; however, most responses targeting BRAFV600E have been often short-lived and drug resistance is inevitable [76]. Thus, exploring the metabolic reprogramming in BRAF mutant cancer is necessary to overcome these issues. In melanoma, activated BRAF promotes metabolic reprogramming by suppression of oxidative phosphorylation through the mitochondrial master regulator PPAR-γ coactivator 1α (PGC1α) [77]. Additionally, a study on slow-cycling, chemotherapy-resistant BRAFV600E-mutated melanoma showed that oxidative phosphorylation enzymes were upregulated, and consequently, their inhibition resulted in cell death [78]. Treatment with an oxidative phosphorylation inhibitor also improved survival and decreased the development of brain metastases in BRAF/MEK inhibitor-resistant mice [79]. In a mouse model of BRAFV600E-driven lung cancer, Atg7 deletion initially provided an advantage in tumor growth due to the induction of oxidative stress. However, at later stages of tumorigenesis, loss of autophagy led to metabolic crisis resulting in defective mitochondria, reduced tumor growth and the conversion of adenomas and adenocarcinomas to oncocytomas [80]. In colorectal cancer, BRAFV600E-mutated tumors expressed high levels of glycolytic enzyme ENO2 and knockdown of ENO2 led to the inhibition of proliferation and migration of BRAFV600E-mutated cancer cells [81]. Inhibition of ENO2 resulted in enhanced sensitivity to vemurafenib, a selective inhibitor of BRAFV600E. Thus, targeting mutation-dependent metabolic dysregulation can counteract drug resistance, providing a unique opportunity for identifying novel therapeutic strategies.
- Metabolic reprogramming in the tumor microenvironment that houses cancerous and immune cells has become an important consideration for cancer treatment. Failure of T cells to protect against cancer is thought to result from lack of antigen recognition, and tumor cells and tumor-infiltrating lymphocytes are known to compete for glucose within the tumor niche [82]. Immune checkpoint blockade antibodies directly altered by tumors have been shown to dampen glycolysis by inhibiting mammalian target of rapamycin (mTOR) activity and decreasing expression of glycolysis enzymes. Moreover, activated cytotoxic CD8+ T cells require metabolic shift from oxidative phosphorylation to aerobic glycolysis to maintain effector function [83], and serine metabolism is suggested as an indispensable part of T cell growth and function [84]. A combination of bioenergetic profiling and 13C-glucose tracing has been used to study the metabolism of CD8+ T cells responding to infection [85]. Physiologically activated CD8+ T cells displayed greater rates of oxidative metabolism, higher bioenergetic capacity, differential use of pyruvate, and prominent flow of 13C-glucose carbon to anabolic pathways, including nucleotide and serine biosynthesis. Quantitative elucidation of metabolic reprogramming during immune responses in cancer patients is an important future objective. Mapping metabolic fluxes in cells that constitute tumor microenvironment and across tissues that are involved in cancer-associated systemic inflammation will render cancers increasingly surmountable.
METABOLISM IN THYROID AND OTHER CANCERS
- The role of insulin in controlling metabolic fluxes has been extensively studied. However, flux control in response to glucocorticoids and thyroid hormones is less well understood. A comprehensive understanding of the metabolic fluxes underlying endocrine diseases is crucial for the development of targeted therapeutic interventions. By elucidating the metabolic alterations in diseases at a systems level, we can identify potential biomarkers for early detection, prognosis, and monitoring of disease progression. Additionally, studying the intricate metabolic networks and pathways involved in endocrine disorders can unveil novel therapeutic targets, enabling the development of personalized treatment strategies that address the underlying metabolic dysregulation.
- Endocrinological disorders frequently have a genetic basis, with mutations in specific genes contributing to their development. Advances in genomics have enabled the identification of numerous genetic variants associated with these disorders. However, the relationship between these genetic variations and the resulting metabolic phenotypes is often intricate and multifaceted. Also, while omics technologies can unveil the molecular aberrations responsible for disruptions in hormone production, secretion, and signaling, the translation of such findings into observable metabolic phenotypes can be challenging due to the intricate nature of hormone regulation. The interplay between genetics, molecular mechanisms, and metabolic outcomes is further complicated by individual variability and environmental influences.
- Aging is a complex biological process characterized by the gradual deterioration of various physiological functions and an increased susceptibility to diseases. The endocrine system plays a critical role in regulating various metabolic processes, and its dysregulation can contribute to age-related metabolic changes. Age-related changes in insulin sensitivity and secretion can disrupt glucose homeostasis [86,87]. Thyroid function tends to decrease with age, leading to a decrease in metabolic rate and potential weight gain and changed metabolic pathways [88,89]. Moreover, dysregulation of cortisol secretion, often seen in chronic stress, can contribute to metabolic disturbances and accelerated aging [90]. Integrative health approaches also play a pivotal role in addressing the challenges of aging by promoting a comprehensive understanding of physical, mental, and emotional well-being, which can be essential for maintaining health as individuals grow older.
- Advances in in vivo MFA using stable isotope tracers have facilitated assessment of differences in metabolic pathway utilization in tissues and quantification of systemic turnover rates of circulating metabolites and their contribution to multiple organs [91-94]. To bridge the gap between omics data and metabolic phenotypes in endocrinological disorders, interdisciplinary approaches are crucial [95]. Integrating omics data with clinical observations, longitudinal studies, and functional assays can provide a more holistic understanding of the disease mechanisms and their metabolic consequences. Additionally, computational modeling and systems biology approaches can help unravel the complex relationships between genetic variations, molecular pathways, and metabolic outcomes. Integrating metabolic network analysis and pathway enrichment tools provides a valuable resource to conduct systems-level analyses, enabling the exploration of metabolic alterations, pathway dysregulations, and the dynamic rewiring of metabolic networks (Table 1) [96-114]. As technology and knowledge advance, researchers are working towards unraveling the intricate connections between genetics, molecular biology, and the clinical realities of endocrinological disorders, ultimately leading to improved diagnostics, targeted therapies, and personalized medicine.
CONCLUSIONS
-
CONFLICTS OF INTEREST
No potential conflict of interest relevant to this article was reported.
Article information
-
Acknowledgements
- This work was supported by the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), the Ministry of Health and Welfare under Award Number HR20C0025, the National Research Foundation of Korea (NRF) under Award Number 2021R1C1C1011183 (Yea Eun Kang), the National Institute of General Medical Sciences of the National Institutes of Health under Award Number R35GM143127 (Junyoung O. Park) and the National Institutes of Health Biotechnology Training Grant under Award Number T32GM067555 (Aliya Lakhani).
Fig. 1.Systems-level metabolic analysis in the context of endocrine disorders and cancer. By integrating advanced analytical techniques such as liquid chromatography-mass spectrometry, magnetic resonance spectroscopy, and flux analyzers and omics methods, we gain a deeper understanding of the complex metabolic alterations underlying endocrine disorders and cancer, paving the way for more effective diagnostic tools and targeted therapies. Created with BioRender.com.
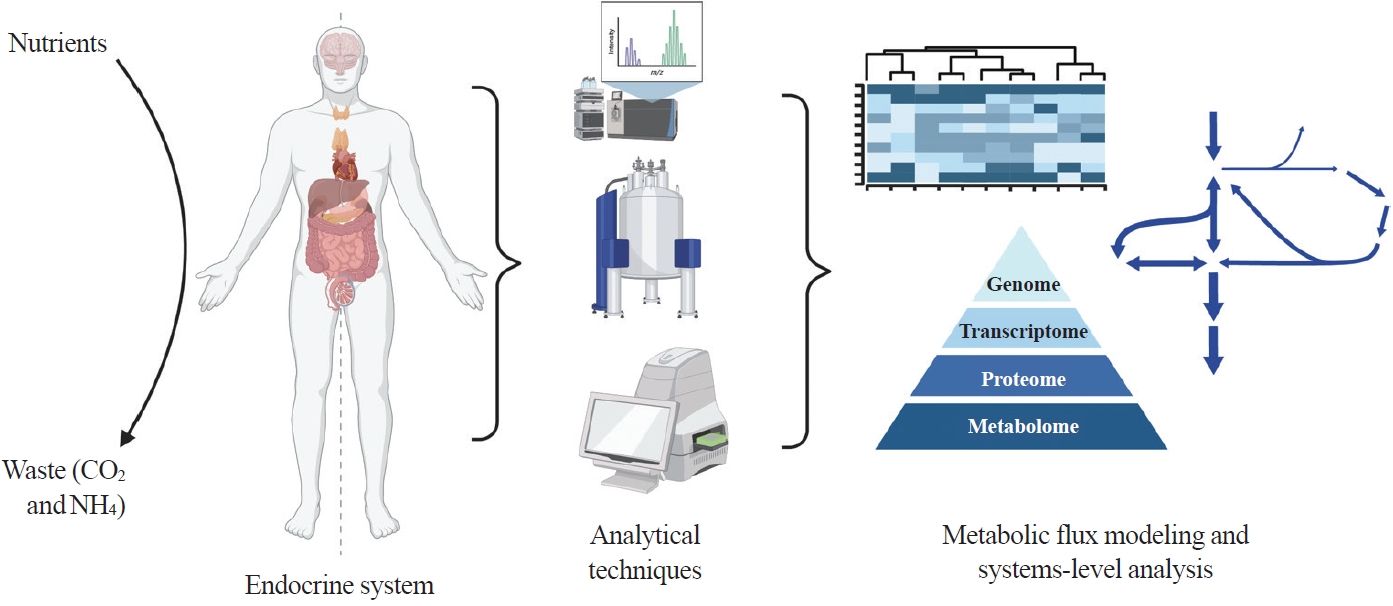
Table 1.Metabolic Networks and Pathway Analysis Tools
| Tool | Applications in endocrine disorders and cancer | |
|---|---|---|
| Metabolic network reconstruction | Metabolic reconstructions (e.g., Recon3D [96]) | Provides a detailed metabolic network model for studying metabolic alterations |
| Supports integration of multi-omics data | ||
| Human Metabolome Database (HMDB [97]) | A valuable resource for annotating metabolomics data and linking metabolites to pathways | |
| KEGG (Kyoto Encyclopedia of Genes and Genomes [98]) | Provides a wide range of metabolic pathway maps and gene annotations | |
| HumanCyc [99] | Provides a comprehensive collection of metabolic pathways and enzymatic reactions in humans | |
| Biochemical Genetic and Genomic (BiGG) models [100] | Offers a repository of curated metabolic models for various organisms | |
| Metabolic network analysis tools | Constraint-Based Reconstruction and Analysis (COBRA) toolbox [101] | Allows constraint-based modeling and flux analysis of metabolic networks |
| Pathway tools [102] | Software for pathway/genome databases and systems biology research that supports the construction and visualization of metabolic pathways | |
| Isotopomer Network Compartmental Analysis (INCA 2.0) [103] | Software that performs steady-state metabolic flux analysis and isotopically non-stationary metabolic flux analysis | |
| Metran [104] | Software for performing 13C-metabolic flux analysis, tracer experiment design, and statistical analysis | |
| 13CFLUX2 [105] | Facilitates the measurement and modeling of carbon fluxes in metabolic pathways in vivo | |
| mfapy [106] | Offers a user-friendly interface for performing flux balance analysis and metabolic modeling, focusing on data analysis procedures | |
| Pathway enrichment analysis | MetaboAnalyst [107] | Facilitates pathway enrichment analysis using metabolomics data |
| Database for Annotation, Visualization, and Integrated Discovery (DAVID) [108] | Useful for gene enrichment analysis to identify overrepresented pathways and functions | |
| Enrichr [109] | Performs gene set enrichment analysis (GSEA) to identify enriched pathways and processes | |
| Metabolomics Pathway Analysis (MetPA) tool [110] | Focuses on the interpretation of metabolomics data in the context of metabolic pathways | |
| Network visualization | Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) [111] | Provides protein-protein interaction networks, helping visualize connections between genes and proteins |
| OmicsNet [112] | Integrates multi-omics data (e.g., genomics, proteomics, metabolomics) and visualizes molecular interactions | |
| Cytoscape [113] | Helps visualize metabolic networks and omics data in a network context | |
| MetExplore [114] | Facilitates the visualization and interpretation of metabolomics data in the context of genome-scale reconstructed metabolic networks |
- 1. J Ryan P, Riechman SE, Fluckey JD, Wu G. Interorgan metabolism of amino acids in human health and disease. Adv Exp Med Biol 2021;1332:129–49.PubMed
- 2. Flynn NE, Shaw MH, Becker JT. Amino acids in health and endocrine function. Adv Exp Med Biol 2020;1265:97–109.ArticlePubMed
- 3. Martinez-Reyes I, Chandel NS. Cancer metabolism: looking forward. Nat Rev Cancer 2021;21:669–80.ArticlePubMedPDF
- 4. Xu X, Peng Q, Jiang X, Tan S, Yang Y, Yang W, et al. Metabolic reprogramming and epigenetic modifications in cancer: from the impacts and mechanisms to the treatment potential. Exp Mol Med 2023;55:1357–70.ArticlePubMedPMCPDF
- 5. Wei Q, Qian Y, Yu J, Wong CC. Metabolic rewiring in the promotion of cancer metastasis: mechanisms and therapeutic implications. Oncogene 2020;39:6139–56.ArticlePubMedPMCPDF
- 6. Moreno-Fernandez S, Garces-Rimon M, Vera G, Astier J, Landrier JF, Miguel M. High fat/high glucose diet induces metabolic syndrome in an experimental rat model. Nutrients 2018;10:1502.ArticlePubMedPMC
- 7. Uematsu S, Ohno S, Tanaka KY, Hatano A, Kokaji T, Ito Y, et al. Multi-omics-based label-free metabolic flux inference reveals obesity-associated dysregulatory mechanisms in liver glucose metabolism. iScience 2022;25:103787.ArticlePubMedPMC
- 8. O’Donovan SD, Lenz M, Vink RG, Roumans NJ, de Kok TM, Mariman EC, et al. A computational model of postprandial adipose tissue lipid metabolism derived using human arteriovenous stable isotope tracer data. PLoS Comput Biol 2019;15:e1007400.ArticlePubMedPMC
- 9. Neinast MD, Jang C, Hui S, Murashige DS, Chu Q, Morscher RJ, et al. Quantitative analysis of the whole-body metabolic fate of branched-chain amino acids. Cell Metab 2019;29:417–29.ArticlePubMedPMC
- 10. Maahs DM, West NA, Lawrence JM, Mayer-Davis EJ. Epidemiology of type 1 diabetes. Endocrinol Metab Clin North Am 2010;39:481–97.ArticlePubMedPMC
- 11. Petersen MC, Vatner DF, Shulman GI. Regulation of hepatic glucose metabolism in health and disease. Nat Rev Endocrinol 2017;13:572–87.ArticlePubMedPMCPDF
- 12. Savolainen O, Fagerberg B, Vendelbo Lind M, Sandberg AS, Ross AB, Bergstrom G. Biomarkers for predicting type 2 diabetes development: can metabolomics improve on existing biomarkers? PLoS One 2017;12:e0177738.ArticlePubMedPMC
- 13. Siddik MA, Shin AC. Recent progress on branched-chain amino acids in obesity, diabetes, and beyond. Endocrinol Metab (Seoul) 2019;34:234–46.ArticlePubMedPMCPDF
- 14. Li X, Wang X, Liu R, Ma Y, Guo H, Hao L, et al. Chronic leucine supplementation increases body weight and insulin sensitivity in rats on high-fat diet likely by promoting insulin signaling in insulin-target tissues. Mol Nutr Food Res 2013;57:1067–79.ArticlePubMed
- 15. Guasch-Ferre M, Hruby A, Toledo E, Clish CB, MartinezGonzalez MA, Salas-Salvado J, et al. Metabolomics in prediabetes and diabetes: a systematic review and meta-analysis. Diabetes Care 2016;39:833–46.ArticlePubMedPMCPDF
- 16. Jang C, Oh SF, Wada S, Rowe GC, Liu L, Chan MC, et al. A branched-chain amino acid metabolite drives vascular fatty acid transport and causes insulin resistance. Nat Med 2016;22:421–6.PubMedPMC
- 17. Blair MC, Neinast MD, Jang C, Chu Q, Jung JW, Axsom J, et al. Branched-chain amino acid catabolism in muscle affects systemic BCAA levels but not insulin resistance. Nat Metab 2023;5:589–606.ArticlePubMedPDF
- 18. Schaller S, Willmann S, Lippert J, Schaupp L, Pieber TR, Schuppert A, et al. A generic integrated physiologically based whole-body model of the glucose-insulin-glucagon regulatory system. CPT Pharmacometrics Syst Pharmacol 2013;2:e65.ArticlePubMedPMCPDF
- 19. Lahoz-Beneytez J, Schaller S, Macallan D, Eissing T, Niederalt C, Asquith B. Physiologically based simulations of deuterated glucose for quantifying cell turnover in humans. Front Immunol 2017;8:474.ArticlePubMedPMC
- 20. Thiele I, Sahoo S, Heinken A, Hertel J, Heirendt L, Aurich MK, et al. Personalized whole-body models integrate metabolism, physiology, and the gut microbiome. Mol Syst Biol 2020;16:e8982.ArticlePubMedPMCPDF
- 21. Ben Guebila M, Thiele I. Dynamic flux balance analysis of whole-body metabolism for type 1 diabetes. Nat Comput Sci 2021;1:348–61.ArticlePubMedPDF
- 22. Paul A, Azhar S, Das PN, Bairagi N, Chatterjee S. Elucidating the metabolic characteristics of pancreatic β-cells from patients with type 2 diabetes (T2D) using a genome-scale metabolic modeling. Comput Biol Med 2022;144:105365.ArticlePubMed
- 23. Chen M, Zheng H, Xu M, Zhao L, Zhang Q, Song J, et al. Changes in hepatic metabolic profile during the evolution of STZ-induced diabetic rats via an 1H NMR-based metabonomic investigation. Biosci Rep 2019;39:BSR20181379.ArticlePubMedPMCPDF
- 24. Rider OJ, Apps A, Miller JJ, Lau JY, Lewis AJ, Peterzan MA, et al. Noninvasive in vivo assessment of cardiac metabolism in the healthy and diabetic human heart using hyperpolarized 13C MRI. Circ Res 2020;126:725–36.ArticlePubMedPMC
- 25. Haythorne E, Rohm M, van de Bunt M, Brereton MF, Tarasov AI, Blacker TS, et al. Diabetes causes marked inhibition of mitochondrial metabolism in pancreatic β-cells. Nat Commun 2019;10:2474.ArticlePubMedPMCPDF
- 26. Rahim M, Nakhe AY, Banerjee DR, Overway EM, Bosma KJ, Rosch JC, et al. Glucose-6-phosphatase catalytic subunit 2 negatively regulates glucose oxidation and insulin secretion in pancreatic β-cells. J Biol Chem 2022;298:101729.ArticlePubMedPMC
- 27. Miller RA, Shi Y, Lu W, Pirman DA, Jatkar A, Blatnik M, et al. Targeting hepatic glutaminase activity to ameliorate hyperglycemia. Nat Med 2018;24:518–24.ArticlePubMedPMCPDF
- 28. Wu Y, Wong CW, Chiles EN, Mellinger AL, Bae H, Jung S, et al. Glycerate from intestinal fructose metabolism induces islet cell damage and glucose intolerance. Cell Metab 2022;34:1042–53.ArticlePubMedPMC
- 29. Perez-Ramirez CA, Nakano H, Law RC, Matulionis N, Thompson J, Pfeiffer A, et al. Atlas of fetal metabolism during mid-to-late gestation and diabetic pregnancy. bioRxiv 2023 Mar 19 [Preprint]. https://doi.org/10.1101/2023.03.16.532852.Article
- 30. Nieman LK. Recent updates on the diagnosis and management of Cushing’s syndrome. Endocrinol Metab (Seoul) 2018;33:139–46.ArticlePubMedPMCPDF
- 31. Giordano R, Guaraldi F, Berardelli R, Karamouzis I, D’Angelo V, Marinazzo E, et al. Glucose metabolism in patients with subclinical Cushing’s syndrome. Endocrine 2012;41:415–23.ArticlePubMedPDF
- 32. Kuo T, McQueen A, Chen TC, Wang JC. Regulation of glucose homeostasis by glucocorticoids. Adv Exp Med Biol 2015;872:99–126.ArticlePubMedPMC
- 33. Kotłowska A, Puzyn T, Sworczak K, Stepnowski P, Szefer P. Metabolomic biomarkers in urine of Cushing’s syndrome patients. Int J Mol Sci 2017;18:294.ArticlePubMedPMC
- 34. Eisenhofer G, Masjkur J, Peitzsch M, Di Dalmazi G, Bidlingmaier M, Gruber M, et al. Plasma steroid metabolome profiling for diagnosis and subtyping patients with Cushing syndrome. Clin Chem 2018;64:586–96.ArticlePubMedPDF
- 35. Murakami M, Sun N, Li F, Feuchtinger A, Gomez-Sanchez C, Fassnacht M, et al. In situ metabolomics of cortisol-producing adenomas. Clin Chem 2023;69:149–59.ArticlePubMedPMCPDF
- 36. Ahn CH, Lee C, Shim J, Kong SH, Kim SJ, Kim YH, et al. Metabolic changes in serum steroids for diagnosing and subtyping Cushing’s syndrome. J Steroid Biochem Mol Biol 2021;210:105856.ArticlePubMed
- 37. Ijare OB, Baskin DS, Pichumani K. Ex vivo 1H NMR study of pituitary adenomas to differentiate various immunohistochemical subtypes. Sci Rep 2019;9:3007.ArticlePubMedPMCPDF
- 38. Ju SH, Lee SE, Kang YE, Shong M. Development of metabolic synthetic lethality and its implications for thyroid cancer. Endocrinol Metab (Seoul) 2022;37:53–61.ArticlePubMedPMCPDF
- 39. Kim JT, Lim MA, Lee SE, Kim HJ, Koh HY, Lee JH, et al. Adrenomedullin2 stimulates progression of thyroid cancer in mice and humans under nutrient excess conditions. J Pathol 2022;258:264–77.ArticlePubMedPMCPDF
- 40. Johnson JM, Lai SY, Cotzia P, Cognetti D, Luginbuhl A, Pribitkin EA, et al. Mitochondrial metabolism as a treatment target in anaplastic thyroid cancer. Semin Oncol 2015;42:915–22.ArticlePubMedPMC
- 41. Abooshahab R, Gholami M, Sanoie M, Azizi F, Hedayati M. Advances in metabolomics of thyroid cancer diagnosis and metabolic regulation. Endocrine 2019;65:1–14.ArticlePubMedPDF
- 42. Tseng CH. Metformin reduces thyroid cancer risk in Taiwanese patients with type 2 diabetes. PLoS One 2014;9:e109852.ArticlePubMedPMC
- 43. Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. BMJ 2005;330:1304–5.ArticlePubMedPMC
- 44. Yu Y, Feng C, Kuang J, Guo L, Guan H. Metformin exerts an antitumoral effect on papillary thyroid cancer cells through altered cell energy metabolism and sensitized by BACH1 depletion. Endocrine 2022;76:116–31.ArticlePubMedPDF
- 45. Bolf EL, Beadnell TC, Rose MM, D’Alessandro A, Nemkov T, Hansen KC, et al. Dasatinib and trametinib promote anti-tumor metabolic activity. Cells 2023;12:1374.ArticlePubMedPMC
- 46. Davidson CD, Tomczak JA, Amiel E, Carr FE. Inhibition of glycogen metabolism induces reactive oxygen species-dependent cytotoxicity in anaplastic thyroid cancer in female mice. Endocrinology 2022;163:bqac169.ArticlePubMedPMCPDF
- 47. Tanner LB, Goglia AG, Wei MH, Sehgal T, Parsons LR, Park JO, et al. Four key steps control glycolytic flux in mammalian cells. Cell Syst 2018;7:49–62.ArticlePubMedPMC
- 48. Abu-Amero KK, Alzahrani AS, Zou M, Shi Y. High frequency of somatic mitochondrial DNA mutations in human thyroid carcinomas and complex I respiratory defect in thyroid cancer cell lines. Oncogene 2005;24:1455–60.ArticlePubMedPDF
- 49. Su X, Wang W, Ruan G, Liang M, Zheng J, Chen Y, et al. A comprehensive characterization of mitochondrial genome in papillary thyroid cancer. Int J Mol Sci 2016;17:1594.ArticlePubMedPMC
- 50. Kurashige T, Shimamura M, Hamada K, Matsuse M, Mitsutake N, Nagayama Y. Characterization of metabolic reprogramming by metabolomics in the oncocytic thyroid cancer cell line XTC.UC1. Sci Rep 2023;13:149.ArticlePubMedPMCPDF
- 51. Metere A, Graves CE, Chirico M, Caramujo MJ, Pisanu ME, Iorio E. Metabolomic reprogramming detected by 1HNMR spectroscopy in human thyroid cancer tissues. Biology (Basel) 2020;9:112.ArticlePubMedPMC
- 52. Lee SE, Park S, Yi S, Lim MA, Chang JW, Won HR, et al. Mitochondrial SHMT2 is a crucial therapeutic target in dedifferentiated thyroid cancer. Res Sq 2022 Aug 2 [Preprint]. https://doi.org/10.21203/rs.3.rs-1881482/v1.Article
- 53. Sugarman AJ, Huynh LD, Shabro A, Di Cristofano A. Anaplastic thyroid cancer cells upregulate mitochondrial onecarbon metabolism to meet purine demand, eliciting a critical targetable vulnerability. Cancer Lett 2023;568:216304.ArticlePubMed
- 54. Lenders JW, Eisenhofer G, Mannelli M, Pacak K. Phaeochromocytoma. Lancet 2005;366:665–75.ArticlePubMed
- 55. Jochmanova I, Yang C, Zhuang Z, Pacak K. Hypoxia-inducible factor signaling in pheochromocytoma: turning the rudder in the right direction. J Natl Cancer Inst 2013;105:1270–83.ArticlePubMedPMC
- 56. Griffin JL. Metabonomics: NMR spectroscopy and pattern recognition analysis of body fluids and tissues for characterisation of xenobiotic toxicity and disease diagnosis. Curr Opin Chem Biol 2003;7:648–54.ArticlePubMed
- 57. Lendvai N, Pawlosky R, Bullova P, Eisenhofer G, Patocs A, Veech RL, et al. Succinate-to-fumarate ratio as a new metabolic marker to detect the presence of SDHB/D-related paraganglioma: initial experimental and ex vivo findings. Endocrinology 2014;155:27–32.ArticlePubMedPMCPDF
- 58. Richter S, Gieldon L, Pang Y, Peitzsch M, Huynh T, Leton R, et al. Metabolome-guided genomics to identify pathogenic variants in isocitrate dehydrogenase, fumarate hydratase, and succinate dehydrogenase genes in pheochromocytoma and paraganglioma. Genet Med 2019;21:705–17.ArticlePubMedPMCPDF
- 59. Richter S, Peitzsch M, Rapizzi E, Lenders JW, Qin N, de Cubas AA, et al. Krebs cycle metabolite profiling for identification and stratification of pheochromocytomas/paragangliomas due to succinate dehydrogenase deficiency. J Clin Endocrinol Metab 2014;99:3903–11.ArticlePubMedPMCPDF
- 60. Imperiale A, Moussallieh FM, Roche P, Battini S, Cicek AE, Sebag F, et al. Metabolome profiling by HRMAS NMR spectroscopy of pheochromocytomas and paragangliomas detects SDH deficiency: clinical and pathophysiological implications. Neoplasia 2015;17:55–65.ArticlePubMedPMC
- 61. Erlic Z, Kurlbaum M, Deutschbein T, Nolting S, Prejbisz A, Timmers H, et al. Metabolic impact of pheochromocytoma/paraganglioma: targeted metabolomics in patients before and after tumor removal. Eur J Endocrinol 2019;181:647–57.ArticlePubMed
- 62. Ku EJ, Lee C, Shim J, Lee S, Kim KA, Kim SW, et al. Metabolic subtyping of adrenal tumors: prospective multicenter cohort study in Korea. Endocrinol Metab (Seoul) 2021;36:1131–41.ArticlePubMedPMCPDF
- 63. Aprile M, Cataldi S, Perfetto C, Federico A, Ciccodicola A, Costa V. Targeting metabolism by B-raf inhibitors and diclofenac restrains the viability of BRAF-mutated thyroid carcinomas with Hif-1α-mediated glycolytic phenotype. Br J Cancer 2023;129:249–65.ArticlePubMedPMCPDF
- 64. Nagarajan A, Malvi P, Wajapeyee N. Oncogene-directed alterations in cancer cell metabolism. Trends Cancer 2016;2:365–77.ArticlePubMedPMC
- 65. Levine AJ, Puzio-Kuter AM. The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science 2010;330:1340–4.ArticlePubMed
- 66. Gaglio D, Metallo CM, Gameiro PA, Hiller K, Danna LS, Balestrieri C, et al. Oncogenic K-Ras decouples glucose and glutamine metabolism to support cancer cell growth. Mol Syst Biol 2011;7:523.ArticlePubMedPMCPDF
- 67. Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012;149:656–70.PubMedPMC
- 68. Amendola CR, Mahaffey JP, Parker SJ, Ahearn IM, Chen WC, Zhou M, et al. KRAS4A directly regulates hexokinase 1. Nature 2019;576:482–6.ArticlePubMedPMCPDF
- 69. Mukhopadhyay S, Vander Heiden MG, McCormick F. The metabolic landscape of RAS-driven cancers from biology to therapy. Nat Cancer 2021;2:271–83.ArticlePubMedPMCPDF
- 70. Mukhopadhyay S, Goswami D, Adiseshaiah PP, Burgan W, Yi M, Guerin TM, et al. Undermining glutaminolysis bolsters chemotherapy while NRF2 promotes chemoresistance in KRAS-driven pancreatic cancers. Cancer Res 2020;80:1630–43.ArticlePubMedPMCPDF
- 71. Grabocka E, Bar-Sagi D. Mutant KRAS enhances tumor cell fitness by upregulating stress granules. Cell 2016;167:1803–13.ArticlePubMedPMC
- 72. Song YS, Lim JA, Park YJ. Mutation profile of well-differentiated thyroid cancer in Asians. Endocrinol Metab (Seoul) 2015;30:252–62.ArticlePubMedPMC
- 73. Park SJ, Kang YE, Kim JH, Park JL, Kim SK, Baek SW, et al. Transcriptomic analysis of papillary thyroid cancer: a focus on immune-subtyping, oncogenic fusion, and recurrence. Clin Exp Otorhinolaryngol 2022;15:183–93.ArticlePubMedPMCPDF
- 74. Kang YE, Hwang B, Lee JH, Shong M, Yi HS, Koo BS, et al. The significance of transcriptomic signatures in the multifocal papillary thyroid carcinoma: two mRNA expression patterns with distinctive clinical behavior from the Cancer Genome Atlas (TCGA) database. Int J Thyroidol 2020;13:1–12.Article
- 75. Gao Y, Yang F, Yang XA, Zhang L, Yu H, Cheng X, et al. Mitochondrial metabolism is inhibited by the HIF1α-MYCPGC-1β axis in BRAF V600E thyroid cancer. FEBS J 2019;286:1420–36.PubMed
- 76. Falchook GS, Millward M, Hong D, Naing A, Piha-Paul S, Waguespack SG, et al. BRAF inhibitor dabrafenib in patients with metastatic BRAF-mutant thyroid cancer. Thyroid 2015;25:71–7.ArticlePubMedPMC
- 77. Haq R, Shoag J, Andreu-Perez P, Yokoyama S, Edelman H, Rowe GC, et al. Oncogenic BRAF regulates oxidative metabolism via PGC1α and MITF. Cancer Cell 2013;23:302–15.ArticlePubMedPMC
- 78. Roesch A, Vultur A, Bogeski I, Wang H, Zimmermann KM, Speicher D, et al. Overcoming intrinsic multidrug resistance in melanoma by blocking the mitochondrial respiratory chain of slow-cycling JARID1B(high) cells. Cancer Cell 2013;23:811–25.ArticlePubMedPMC
- 79. Fischer GM, Jalali A, Kircher DA, Lee WC, McQuade JL, Haydu LE, et al. Molecular profiling reveals unique immune and metabolic features of melanoma brain metastases. Cancer Discov 2019;9:628–45.PubMedPMC
- 80. Strohecker AM, Guo JY, Karsli-Uzunbas G, Price SM, Chen GJ, Mathew R, et al. Autophagy sustains mitochondrial glutamine metabolism and growth of BrafV600E-driven lung tumors. Cancer Discov 2013;3:1272–85.ArticlePubMedPMCPDF
- 81. Yukimoto R, Nishida N, Hata T, Fujino S, Ogino T, Miyoshi N, et al. Specific activation of glycolytic enzyme enolase 2 in BRAF V600E-mutated colorectal cancer. Cancer Sci 2021;112:2884–94.ArticlePubMedPMCPDF
- 82. Chang CH, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell 2015;162:1229–41.ArticlePubMedPMC
- 83. Triozzi PL, Stirling ER, Song Q, Westwood B, Kooshki M, Forbes ME, et al. Circulating immune bioenergetic, metabolic, and genetic signatures predict melanoma patients’ response to anti-PD-1 immune checkpoint blockade. Clin Cancer Res 2022;28:1192–202.ArticlePubMedPMCPDF
- 84. Kurniawan H, Franchina DG, Guerra L, Bonetti L, SorianoBaguet L, Grusdat M, et al. Glutathione restricts serine metabolism to preserve regulatory T cell function. Cell Metab 2020;31:920–36.ArticlePubMedPMC
- 85. Ma EH, Verway MJ, Johnson RM, Roy DG, Steadman M, Hayes S, et al. Metabolic profiling using stable isotope tracing reveals distinct patterns of glucose utilization by physiologically activated CD8+ T cells. Immunity 2019;51:856–70.ArticlePubMed
- 86. Karakelides H, Irving BA, Short KR, O’Brien P, Nair KS. Age, obesity, and sex effects on insulin sensitivity and skeletal muscle mitochondrial function. Diabetes 2010;59:89–97.ArticlePubMedPMCPDF
- 87. Chia CW, Egan JM, Ferrucci L. Age-related changes in glucose metabolism, hyperglycemia, and cardiovascular risk. Circ Res 2018;123:886–904.ArticlePubMedPMC
- 88. Hong Y, Kim HJ, Park S, Yi S, Lim MA, Lee SE, et al. Single cell analysis of human thyroid reveals the transcriptional signatures of aging. Endocrinology 2023;164:bqad029.ArticlePubMedPDF
- 89. Lee J, Yi S, Kang YE, Kim HW, Joung KH, Sul HJ, et al. Morphological and functional changes in the thyroid follicles of the aged murine and humans. J Pathol Transl Med 2016;50:426–35.ArticlePubMedPMCPDF
- 90. Martocchia A, Stefanelli M, Falaschi GM, Toussan L, Ferri C, Falaschi P. Recent advances in the role of cortisol and metabolic syndrome in age-related degenerative diseases. Aging Clin Exp Res 2016;28:17–23.ArticlePubMedPDF
- 91. Bednarski TK, Rahim M, Young JD. In vivo2H/13C flux analysis in metabolism research. Curr Opin Biotechnol 2021;71:1–8.ArticlePubMedPMC
- 92. Fernandez-Garcia J, Altea-Manzano P, Pranzini E, Fendt SM. Stable isotopes for tracing mammalian-cell metabolism in vivo. Trends Biochem Sci 2020;45:185–201.ArticlePubMed
- 93. Hasenour CM, Rahim M, Young JD. In vivo estimates of liver metabolic flux assessed by 13C-propionate and 13Clactate are impacted by tracer recycling and equilibrium assumptions. Cell Rep 2020;32:107986.ArticlePubMedPMC
- 94. Hui S, Cowan AJ, Zeng X, Yang L, TeSlaa T, Li X, et al. Quantitative fluxomics of circulating metabolites. Cell Metab 2020;32:676–88.ArticlePubMedPMC
- 95. Law RC, Lakhani A, O’Keeffe S, Ersan S, Park JO. Integrative metabolic flux analysis reveals an indispensable dimension of phenotypes. Curr Opin Biotechnol 2022;75:102701.ArticlePubMedPMC
- 96. Brunk E, Sahoo S, Zielinski DC, Altunkaya A, Drager A, Mih N, et al. Recon3D enables a three-dimensional view of gene variation in human metabolism. Nat Biotechnol 2018;36:272–81.ArticlePubMedPMCPDF
- 97. Wishart DS, Tzur D, Knox C, Eisner R, Guo AC, Young N, et al. HMDB: the Human Metabolome Database. Nucleic Acids Res 2007;35(Database issue):D521–6.ArticlePubMedPMC
- 98. Kanehisa M, Goto S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res 2000;28:27–30.ArticlePubMedPMC
- 99. Romero P, Wagg J, Green ML, Kaiser D, Krummenacker M, Karp PD. Computational prediction of human metabolic pathways from the complete human genome. Genome Biol 2005;6:R2.ArticlePubMedPMC
- 100. Schellenberger J, Park JO, Conrad TM, Palsson BO. BiGG: a Biochemical Genetic and Genomic knowledgebase of large scale metabolic reconstructions. BMC Bioinformatics 2010;11:213.ArticlePubMedPMCPDF
- 101. Heirendt L, Arreckx S, Pfau T, Mendoza SN, Richelle A, Heinken A, et al. Creation and analysis of biochemical constraint-based models using the COBRA Toolbox v.3.0. Nat Protoc 2019;14:639–702.ArticlePubMedPMCPDF
- 102. Karp PD, Paley S, Romero P. The pathway tools software. Bioinformatics 2002;18 Suppl 1:S225–32.ArticlePubMedPDF
- 103. Rahim M, Ragavan M, Deja S, Merritt ME, Burgess SC, Young JD. INCA 2.0: a tool for integrated, dynamic modeling of NMR- and MS-based isotopomer measurements and rigorous metabolic flux analysis. Metab Eng 2022;69:275–85.ArticlePubMedPMC
- 104. Yoo H, Antoniewicz MR, Stephanopoulos G, Kelleher JK. Quantifying reductive carboxylation flux of glutamine to lipid in a brown adipocyte cell line. J Biol Chem 2008;283:20621–7.ArticlePubMedPMC
- 105. Weitzel M, Noh K, Dalman T, Niedenfuhr S, Stute B, Wiechert W. 13CFLUX2: high-performance software suite for (13)C-metabolic flux analysis. Bioinformatics 2013;29:143–5.ArticlePubMedPMCPDF
- 106. Matsuda F, Maeda K, Taniguchi T, Kondo Y, Yatabe F, Okahashi N, et al. mfapy: an open-source Python package for 13C-based metabolic flux analysis. Metab Eng Commun 2021;13:e00177.ArticlePubMedPMC
- 107. Xia J, Psychogios N, Young N, Wishart DS. MetaboAnalyst: a web server for metabolomic data analysis and interpretation. Nucleic Acids Res 2009;37(Web Server issue):W652–60.ArticlePubMedPMC
- 108. Dennis G Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, et al. DAVID: database for annotation, visualization, and integrated discovery. Genome Biol 2003;4:P3.ArticlePubMedPDF
- 109. Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 2013;14:128.ArticlePubMedPMCPDF
- 110. Xia J, Wishart DS. MetPA: a web-based metabolomics tool for pathway analysis and visualization. Bioinformatics 2010;26:2342–4.ArticlePubMedPDF
- 111. Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res 2019;47(D1):D607–13.ArticlePubMedPMC
- 112. Zhou G, Xia J. OmicsNet: a web-based tool for creation and visual analysis of biological networks in 3D space. Nucleic Acids Res 2018;46(W1):W514–22.ArticlePubMedPMC
- 113. Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 2003;13:2498–504.ArticlePubMedPMC
- 114. Cottret L, Wildridge D, Vinson F, Barrett MP, Charles H, Sagot MF, et al. MetExplore: a web server to link metabolomic experiments and genome-scale metabolic networks. Nucleic Acids Res 2010;38(Web Server issue):W132–7.ArticlePubMedPMC
References
Figure & Data
References
Citations
Citations to this article as recorded by 
- Editorial: Tumor metabolism and programmed cell death
Dan-Lan Pu, Qi-Nan Wu
Frontiers in Endocrinology.2024;[Epub] CrossRef
 PubReader
PubReader ePub Link
ePub Link Cite
Cite