Articles
- Page Path
- HOME > Endocrinol Metab > Volume 39(1); 2024 > Article
-
Review ArticleAdrenal gland The Fascinating Interplay between Growth Hormone, Insulin-Like Growth Factor-1, and Insulin
 Keypoint
Keypoint
Growth hormone, IGF-1, and insulin sometimes antagonize or amplify each other’s direct actions. In this interplay, for translating growth hormone actions into IGF-1 production, the production of endogenous insulin that reaches the liver via the portal vein is an essential factor. This review addresses the interplay between GH, IGF-1, and insulin in type 1 and 2 diabetes mellitus, liver cirrhosis, and acromegaly. -
Eline C. Nijenhuis-Noort, Kirsten A. Berk, Sebastian J. C. M. M. Neggers, Aart J. van der Lely

-
Endocrinology and Metabolism 2024;39(1):83-89.
DOI: https://doi.org/10.3803/EnM.2024.101
Published online: January 9, 2024
Division of Endocrinology, Department of Medicine, Erasmus University Medical Center, Rotterdam, The Netherlands
- Corresponding author: Aart J. van der Lely. Division of Endocrinology, Department of Medicine, Erasmus University Medical Center, Dr. Molewaterplein 40, 3015 GD Rotterdam, The Netherlands Tel: +31-107032861, Fax: +31-107033639, E-mail: a.vanderlelij@erasmusmcnl
• Received: October 30, 2023 • Revised: November 23, 2023 • Accepted: December 6, 2023
Copyright © 2024 Korean Endocrine Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- ABSTRACT
- INTRODUCTION
- HOW DO GH, IGF-1, AND INSULIN CHANGE EACH OTHER’S CONCENTRATIONS?
- THE GH, IGF-1, AND INSULIN INTERPLAY IN TYPE 1 DIABETES MELLITUS
- THE GH, IGF-1, AND INSULIN INTERPLAY IN TYPE 2 DIABETES MELLITUS AND OBESITY
- THE GH, IGF-1, AND INSULIN INTERPLAY IN LIVER FAILURE
- THE GH, IGF-1, AND INSULIN INTERPLAY IN ACROMEGALY
- CONCLUSIONS
- Article information
- References
ABSTRACT
- This review intends to provide the reader with a practical overview of several (patho)physiological conditions in which knowledge of the interplay between growth hormone (GH), insulin-like growth factor-1 (IGF-1), and insulin is important. This might help treating physicians in making the right decisions on how to intervene and improve metabolism for the benefit of patients, and to understand why and how metabolism responds in their specific cases. We will specifically address the interplay between GH, IGF-1, and insulin in type 1 and 2 diabetes mellitus, liver cirrhosis, and acromegaly as examples in which this knowledge is truly necessary.
- Some of the most well-known endocrinological conditions are growth hormone deficiency (GHD), acromegaly, and type 1 and type 2 diabetes mellitus (T1DM and T2DM, respectively). While growth hormone (GH) and serum insulin-like growth factor-1 (IGF-1) are used as parameters of control and efficacy of treatment in GHD and acromegaly, glycosylated hemoglobin (HbA1c) levels are used in T1DM [1-4]. Without exaggeration, treatment is rather oversimplified in all three of these conditions: GHD is treated by GH and the necessary dose is the one that normalizes serum IGF-1. Acromegaly is treated by reducing GH levels or GH actions to the level by which IGF-1 normalizes, and T1DM is treated by insulin until HbA1c levels normalize [1-4]. As GH, IGF-1, and insulin continuously modify each other’s production and actions, a good knowledge of the mechanisms behind these interactions is mandatory.
- This paper does not pretend to provide the reader with a complete overview of the GH, IGF-1, and insulin interplay, including its exceptions and the exceptions to the exceptions. It does, however, intend to provide a practical overview of several (patho)physiological conditions in which knowledge of this interplay is important, to understand why and how metabolism responds. T1DM, T2DM, liver cirrhosis, and acromegaly will be used below as examples in which this knowledge is truly necessary.
INTRODUCTION
- Gahete et al. [5] previously evaluated the separate roles of insulin and IGF-1 in directly regulating GH secretion, using the Cre/loxP system to knock down the insulin receptor (INSR) and IGF-1 receptor (IGFIR) in primary mouse pituitary cell cultures. They found that the insulin-mediated suppression of GH is independent of the IGFIR [5]. In addition, pharmacological blockade of intracellular signals in both mouse and baboon cultures revealed that insulin requires different pathways from IGF-1 to exert a maximal inhibitory effect on GH expression/release [5].
- GH activates GH receptors (GHRs), which activate IGF-1 in the liver through a cascade of processes [6]. The GH/IGF-1 axis plays an important role in the regulation of metabolism. Insufficient GH secretion results in short stature in childhood, while adult (onset) GH deficiency (AGHD) is GHD that occurs in adulthood [6]. Sirtuin 1 (SIRT1) negatively regulates GH-induced IGF-1 production in the liver by directly inhibiting signal transducer and activator of transcription 5 (STAT5) activation, which causes GH resistance under prolonged fasting, starvation, and malnutrition [6].
- In GHD, women require a greater replacement dose of GH than men. This is because androgens enhance, while estrogens attenuate GH action [7]. The estrogen effect is route-dependent, occurring with oral delivery blunting the liver-mediated actions of GH by directly inhibiting GHR signaling.
- Insulin modulates the biological actions of GH by controlling the expression of human hepatic GHRs [8]. Using the human hepatoma cell line HuH7 as a model, Leung et al. [8] investigated insulin regulation of total, intracellular, and cell surface GHRs and receptor biosynthesis and turnover. Insulin upregulated total and intracellular GHRs in a concentration-dependent manner. It increased surface GHRs in a biphasic manner, with a peak response at 10 nmol/L, and it modulated GH-induced Janus kinase-2 phosphorylation in parallel with the expression of surface GHRs [8]. Apparently, insulin regulates hepatic GHR biosynthesis and surface translocation in a reciprocal manner, with surface receptor availability as the net result of the divergent effects. The divergent actions of insulin appear to be mediated by the mitogen-activated protein kinase and phosphatidylinositol 3-kinase pathways, respectively [8].
- The role of IGF-1 feedback regulation in GH production has been demonstrated by pharmacologic interventions and in genetically modified mouse models. In a relatively recent review by Al-Samerria and Radovick [9], the role of IGF-1 in the regulation of the GH-axis was addressed.
- Metabolically, GH promotes anabolic action in most tissues except adipose, where its catabolic action causes the breakdown of stored triglycerides into free fatty acids (FFA). GH antagonizes insulin action via various molecular pathways [10]. Chronic GH secretion suppresses the anti-lipolytic action of insulin and increases FFA flux into the systemic circulation, thereby promoting lipotoxicity, which causes pathophysiological problems, including insulin resistance [10].
- The activity of IGF-1 is in part regulated by its binding proteins. There are six high-affinity binding proteins (insulin-like growth factor-binding protein [IGFBP]-1 to IGFBP-6) that are unrelated to the cell surface receptors [11]. In the circulation, two of the IGFBPs, IGFBP-3 and IGFBP-5, are bound to a further large glycoprotein, the acid-labile subunit, that is present in excess [11]. This ternary complex slows even higher clearance, such that in adult humans, the total IGF-1 concentration in the circulation is around 100 nM. This is approximately 1,000 times higher than the concentration of insulin that does not have a binding protein [11].
- Both insulin and IGF-1 can bind to and activate each other’s receptors, albeit with reduced affinity [12]. When portal insulin levels are low (e.g., during fasting and in T1DM), the low insulin activity in the liver must be able to lower IGF-1 secretion and reduce its activity to prevent IGF-1-induced hypoglycemia by binding to the INSR, as the IGF-1 concentrations are significantly higher—enough to compensate for the lower affinity of IGF-1 to the INSR. As explained above, insulin does that by controlling the expression of human hepatic GHRs [8].
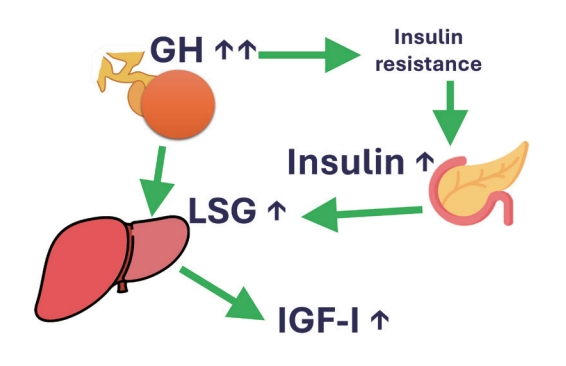
- In conclusion, GH stimulates IGF-1 levels and insulin concentrations, the latter mainly by inducing insulin resistance. IGF-1 decreases GH levels by growth hormone releasing hormone (GHRH)-dependent feedback mechanisms. Insulin increases liver GHR expression, making the liver more GH-sensitive, leading to an increase in IGF-1 and a decrease in GH. On the other hand, low portal insulin levels (e.g., during prolonged fasting) reduce hepatic GHR expression, thereby reducing IGF-1 levels and increasing GH concentrations by the lack of IGF-1 feedback on pituitary GH secretion (Fig. 1).
HOW DO GH, IGF-1, AND INSULIN CHANGE EACH OTHER’S CONCENTRATIONS?
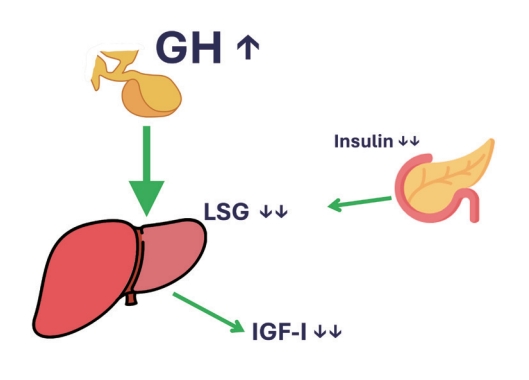
- To understand the dynamics of this interplay in T1DM, one must go back to the relation between portal insulin and GHR expression on hepatocytes, which was so cleverly addressed by Leung et al. [8] and described above. As the main problem lies in the lack of endogenous insulin production, all T1DM subjects will have low portal insulin levels, even when treated with subcutaneous insulin, as this will only increase systemic insulin concentrations. Shishko et al. [13] performed a brilliant study in 36 T1DM patients (ranging in age from 18 to 22 years), within 1 to 3 weeks after diagnosis. T1DM is associated with decreased IGF-1 levels, GH, and IGFBP-1. Since the liver is the major source of IGF and IGFBP production, they examined whether levels of IGFs (IGF-1 and IGF-11) and IGFBPs (IGFBP-1 and IGFBP-3) differed when insulin was infused into the portal or peripheral vascular system [13]. As expected, IGF-1 levels were low before insulin therapy administration [13]. With insulin treatment, IGF-1 levels rose to the normal range and IGF-1 normalization depended on diabetes control and the route of insulin infusion. Diabetic patients with conventional subcutaneous insulin therapy had lower IGF-1 levels than patients with continuous subcutaneous insulin infusion and intraportal insulin infusion [13]. Their results clearly show the role of glycemic control and the route of insulin administration in the normalization of IGF-1, IGFBP-1, and GH in patients with recent-onset T1DM. Only when insulin was given back to where it was missing, the portal vein, did serum IGF-1 levels normalize. This normalization in IGF-1 in turn resulted in a normalization of the elevated GH levels. Therefore, in T1DM, because of the lack of portal insulin levels, the liver stops producing enough IGF-1, which is followed by a compensatory rise in GH levels. These levels can still induce gluconeogenesis and lipolysis.
- In fact, the livers of T1DM patients behave as being in the fasting state as they do not encounter insulin in the portal vein, but still encounter nutrients from the gut that reach them via the portal vein. The lack of insulin overrides the presence of nutrients as a signal, so these livers start to produce glucose via gluconeogenesis and lipolysis while the patient is eating. This condition is generally treated by increasing their systemic insulin levels by subcutaneous (i.e., systemic) injections and not by delivery via the portal system (due to understandable practical reasons) [13,14].
- In this light, we focus on one of the complications of chronically uncontrolled hyperglycemia: end-stage kidney disease, which necessitates kidney replacement therapy. During times of transition to hemodialysis or peritoneal dialysis, consideration must be given to insulin dosing adjustments for subjects with T1DM in efforts to maintain glycemic control. In a recent review by Blaine et al. [15], it was concluded that during peritoneal dialysis, insulin may be administered subcutaneously, intraperitoneally, or with the dialysis fluid. Administration of insulin with dialysate may necessitate a dose increase of up to 30% [15]. Providing the liver with insulin in the dialysate will restore the GHR expression in the liver. That will enable GH to restore IGF-1 production, which will exert feedback on the GH production. Lower GH levels will make the body more insulin-sensitive. This increase in insulin sensitivity will reduce the mandatory dose of insulin for proper glycemic control.
- In conclusion, T1DM patients have absent insulin levels in the portal vein, which makes their liver very GH-resistant. This results in an IGF-1 deficiency and high GH levels in the range seen in acromegaly patients. Treating them with systemic insulin will reduce glucose levels but will not reduce the GH resistance of the liver. Only the administration of insulin directly into the portal vein will really treat the metabolic derangement present in T1DM (Fig. 2).
THE GH, IGF-1, AND INSULIN INTERPLAY IN TYPE 1 DIABETES MELLITUS
- An entirely different situation is present in T2DM and obesity. In that situation, portal insulin levels are generally high due to insulin resistance [16,17]. However, despite the considerable body of evidence pointing to a possible relationship between the state of the adipose tissue depots and regulation of the somatotropic axis, to date the relationship between obesity and low GH status remains incompletely understood [18]. Low GH status in obesity is mainly considered a functional condition, largely reversible after sustained weight loss [18]. Since GH is a diabetogenic molecule, it would not be predicted to be used as a pharmaceutical to treat T2DM. Nonetheless, GH’s lipolytic and anti-lipogenic actions could have potential positive outcomes in T2DM [19,20]. In this regard, GH has been used only to a limited extent in the treatment of patients with T2DM; however, two reports have documented beneficial effects on glucose metabolism when GH treatment was combined with caloric restriction [19,20].
- Although the beneficial effects of tight glycemic control in terms of microvascular complications in patients with T2DM are well established [21], weight gain appears to be a barrier to achieving glycemic targets in diabetes patients. Weight gain is particularly present with the use of thiazolidinediones and insulin secretagogues [22]. They both increase endogenous insulin (signaling) that will make the liver more GH-sensitive (see Leung et al. [8] above). A more sensitive liver needs less GH to produce enough IGF-1. Therefore, the above-mentioned antidiabetic medications will lead to a disbalance in the GH/insulin ratio, meaning a low GH and a high insulin level. In this situation, adipocytes encountering high insulin and low GH levels can only store nutrients and not start lipolysis, the ideal situation for gaining weight instead of losing it.
- In conclusion, whatever medication that increases endogenous portal insulin (signaling) in T2DM patients, regardless of the glucose level (specifically sulfonylurea derivatives and peroxisome proliferator–activated receptor γ [PPAR-γ] agonists, but not dipeptidyl peptidase-4 inhibitor [DPP4i], or glucagonlike peptide-1 [GLP-1] analogues) will lead to an increase in body weight, even when caloric intake is not increased. This is the result of the very high GH sensitivity of a liver expressing high levels of GHRs. Because of the high portal insulin levels, not much GH is needed for IGF-1 production. The low GH to high insulin ratio disables adipose tissues from removing their high lipid content. Therefore, only insulin-sensitizing anti-diabetic drugs are the rational solution for treating T2DM (Fig. 3).
THE GH, IGF-1, AND INSULIN INTERPLAY IN TYPE 2 DIABETES MELLITUS AND OBESITY
- Patients with chronic liver disease are prone to developing sarcopenia, which is defined as loss of skeletal muscle mass, strength, and function, and is associated with increased morbidity and mortality [23]. The pathogenesis of sarcopenia is complex and multifaceted: the chronic catabolic condition, increased energy expenditure, reduced appetite, side effects of multiple therapies, alterations in circulating levels of hormones, low protein synthesis, and the presence of ascites or portosystemic shunts are all factors contributing to muscle atrophy in cirrhotic patients [23]. When the total mass of hepatocytes becomes too small to produce enough IGF-1 and store enough glycogen, glycemic control can become very problematic [24]. When glucose levels tend to decrease, insulin secretion will go down as well. The decreased levels of portal insulin, provided there is still portal flow at all in end-stage cirrhosis, make the remaining hepatocytes even more GH-resistant [8]. This will inevitably result in a severe IGF-1 deficiency, together with high GH levels, very far into the acromegaly range [25]. The high GH levels, together with the often severe IGF-1 deficiency, will lead to a severe catabolic state, including significant lipolysis and proteolysis, comparable to extended fasting during a hunger strike, for instance [26].
- Therefore, IGF-1 deprivation seriously contributes to the progressive malnutrition of cirrhotic patients, increasing the vulnerability of the liver to an inflammatory and oxidative microenvironment with mitochondrial dysfunction. Several studies in animals and humans have been done to test the replacement of IGF-1 as a possible therapeutic option, with promising results [27].
- In conclusion, liver failure results in severe IGF-1 deprivation and elevated GH levels. This will increase catabolism, which is characteristic of liver failure. Restoring IGF-1 by administering it seems to have promising effects, at least in rodent studies, but large-scale human data are missing to date (Fig. 4).
THE GH, IGF-1, AND INSULIN INTERPLAY IN LIVER FAILURE
- Acromegaly is a rare disease, most often caused by a GH-producing tumor of the anterior pituitary [28]. All available (medical) treatment modalities to date are aimed at normalizing serum IGF-1 levels via reduction of either GH overproduction or GH actions [29-32]. The obvious advantage is that efficacy of all treatment modalities can be compared by calculating the percentage of patients that show a drop in their IGF-1 levels to within the age and sex-adjusted normal ranges. This also concerns comparisons between the efficacy of long-acting somatostatin therapies and the GH-receptor antagonist pegvisomant. However, there is one major drawback in this approach, which has significant implications for patient care and future directions in clinical research. It completely ignores the fact that GH and IGF-1 are important factors in the metabolic control of glucose and insulin and vice versa [33].
- Acromegaly patients have elevated IGF-1 levels because they have pituitary tumors that produce too much GH [28]. Of course, this is true, but there is more to this that worsens the situation significantly in untreated acromegaly. In active acromegaly, many observations show that due to the high GH levels, glucose levels tend to be elevated, which is accompanied by the well-known high insulin levels in untreated acromegaly patients [33,34].
- This GH-driven hyperinsulinemia results in a very unwanted situation where the liver becomes even more GH-sensitive. This hypersensitivity for GH further pushes up the already elevated IGF-1 production, while on top of that IGF-1 actions are also increased because of the hyperinsulinemia-induced reduction in IGFBP-1 [35]. The strength of this insulin-driven hypersecretion of IGF-1 was shown in the past by the observation that hypoinsulinemia induced by prolonged fasting can completely normalize serum IGF-1 levels in acromegaly patients [36,37]. What makes the story even more complicated is that GH has concentration-dependent effects on tissues, as there are indications that one needs less GH to stimulate gluconeogenesis and glycolysis versus IGF-1 generation [34,38]. The relatively high systemic insulin levels counteract the lipolytic effects of the high GH levels in fat and muscle, but they do not reach adequate levels in the portal vein, so the liver remains under the control of GH, which still induces gluconeogenesis, increasing hyperglycemia [33].
- In conclusion, acromegaly patients have elevated serum IGF-1 levels because of the pathological hypersecretion of GH by the pituitary tumor. However, because of the concomitant hyperinsulinemia, the liver becomes inadequately hypersensitive to GH, which further worsens the situation. Finally, the actions of the elevated IGF-1 levels are increased because of the drop in IGFBP-1 (Fig. 5).
THE GH, IGF-1, AND INSULIN INTERPLAY IN ACROMEGALY
- GH is not just a hormone that increases IGF-1 concentrations. GH, IGF-1, and insulin sometimes antagonize or amplify each other’s direct actions. In this interplay, for translating GH actions into IGF-1 production, the production of endogenous insulin that reaches the liver via the portal vein is an essential factor. In fact, insulin decides whether GH is allowed to generate IGF-1 production and release by the liver. By not considering GH, IGF-1, and insulin altogether, clinicians will not be able to assess and understand the complex changes in metabolism in which GH, IGF-1, and insulin play a major role. Therefore, in cases of diabetes (both T1DM and T2DM), acromegaly and liver failure, a thorough understanding of normal physiology and pathophysiology will certainly help the treating physician to make the right decisions on how to intervene and improve metabolism for the benefit of patients.
CONCLUSIONS
-
CONFLICTS OF INTEREST
No potential conflict of interest relevant to this article was reported.
Article information
Fig. 1.The positive effect of insulin on the liver sensitivity for growth hormone (LSG). In this way, portal vein insulin levels can control insulin-like growth factor-1 (IGF-1) generation by growth hormone (GH).
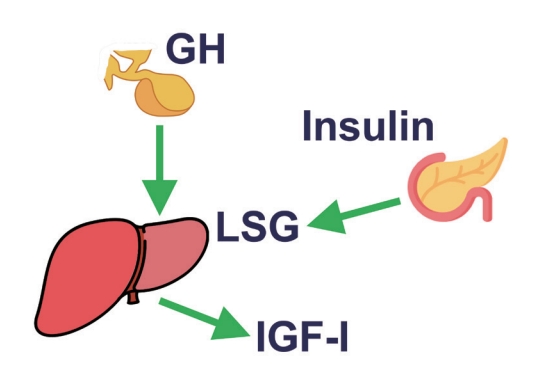
Fig. 2.In type 1 diabetes mellitus, portal vein insulin levels are extremely low. This significantly decreases liver sensitivity for growth hormone (LSG). Therefore, insulin-like growth factor-1 (IGF-1) levels drop, and due to the lack of negative feedback by IGF-1 on the hypothalamus, growth hormone (GH) levels increase significantly.
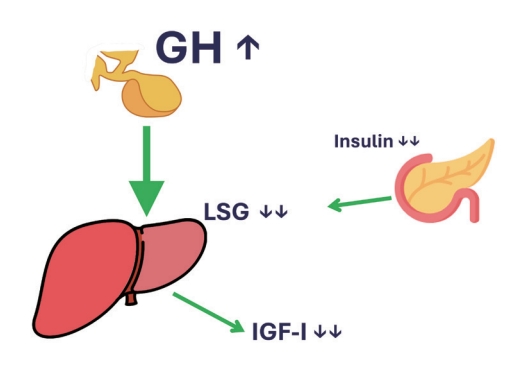
Fig. 3.In type 2 diabetes mellitus, portal vein insulin levels are elevated to compensate for the insulin-resistant state of the body. This significantly increases liver sensitivity for growth hormone (LSG). Therefore, insulin-like growth factor-1 (IGF-1) levels are high in the normal range. Due to the negative feedback by IGF-1 on the hypothalamus, growth hormone (GH) levels decrease significantly. The low GH/insulin ratio will stimulate lipogenesis and inhibit lipolysis. The net result is the tendency to increase body weight.
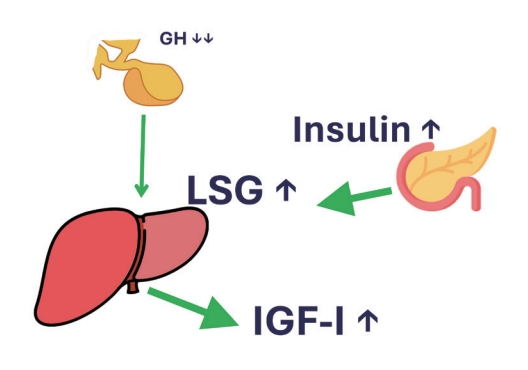
Fig. 4.In liver insufficiency, the liver fails to produce enough insulin-like growth factor-1 (IGF-1). The storage of glycogen is also hampered. Therefore, portal vein insulin levels are low. This decreases liver sensitivity for growth hormone (LSG). Therefore, IGF-1 levels further drop and, due to the lack of negative feedback by IGF-1 on the hypothalamus, growth hormone (GH) levels increase significantly. These GH levels result in an often severe state of catabolism, with significant proteolysis and lipolysis.

Fig. 5.In acromegaly, the somatotropinoma in the anterior pituitary produces excessive amounts of growth hormone (GH). This induces severe insulin resistance. Therefore, portal vein insulin levels are high. High portal insulin concentrations significantly increase liver sensitivity for growth hormone (LSG). This only makes the situation worse, as the high GH levels only further increase the already elevated insulin-like growth factor-1 (IGF-1) concentrations. Thus, in acromegaly, IGF-1 levels are elevated because of the somatotropinoma, but also because of the elevated insulin levels in the portal vein.
- 1. Redondo MJ, Morgan NG. Heterogeneity and endotypes in type 1 diabetes mellitus. Nat Rev Endocrinol 2023;19:542–54.ArticlePubMedPDF
- 2. Midyett LK. One size fits all versus individualized medicine in type 1 diabetes management. Diabetes Technol Ther 2023;25(S3):S42–7.ArticlePubMed
- 3. Yuen KC, Johannsson G, Ho KK, Miller BS, Bergada I, Rogol AD. Diagnosis and testing for growth hormone deficiency across the ages: a global view of the accuracy, caveats, and cut-offs for diagnosis. Endocr Connect 2023;12:e220504.ArticlePubMedPMC
- 4. Kerbel J, Cano-Zaragoza A, Espinosa-Dorado R, Garcia de la Torre KE, Mercado M. Real world data on the epidemiology, diagnosis, and treatment of acromegaly: a registries-based approach. Arch Med Res 2023;54:102856.ArticlePubMed
- 5. Gahete MD, Cordoba-Chacon J, Lin Q, Bruning JC, Kahn CR, Castano JP, et al. Insulin and IGF-I inhibit GH synthesis and release in vitro and in vivo by separate mechanisms. Endocrinology 2013;154:2410–20.ArticlePubMedPMCPDF
- 6. Yamamoto M, Bando H. A new insight into GH regulation and its disturbance from nutrition and autoimmune perspectives. Endocr J 2023;70:867–74.ArticlePubMed
- 7. Ho KK, O’Sullivan AJ, Burt MG. The physiology of growth hormone (GH) in adults: translational journey to GH replacement therapy. J Endocrinol 2023;257:e220197.ArticlePubMed
- 8. Leung KC, Doyle N, Ballesteros M, Waters MJ, Ho KK. Insulin regulation of human hepatic growth hormone receptors: divergent effects on biosynthesis and surface translocation. J Clin Endocrinol Metab 2000;85:4712–20.ArticlePubMed
- 9. Al-Samerria S, Radovick S. The role of insulin-like growth factor-1 (IGF-1) in the control of neuroendocrine regulation of growth. Cells 2021;10:2664.ArticlePubMedPMC
- 10. Sharma R, Kopchick JJ, Puri V, Sharma VM. Effect of growth hormone on insulin signaling. Mol Cell Endocrinol 2020;518:111038.ArticlePubMedPMC
- 11. Firth SM, Baxter RC. Cellular actions of the insulin-like growth factor binding proteins. Endocr Rev 2002;23:824–54.ArticlePubMed
- 12. Taniguchi CM, Emanuelli B, Kahn CR. Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol 2006;7:85–96.ArticlePubMedPDF
- 13. Shishko PI, Dreval AV, Abugova IA, Zajarny IU, Goncharov VC. Insulin-like growth factors and binding proteins in patients with recent-onset type 1 (insulin-dependent) diabetes mellitus: influence of diabetes control and intraportal insulin infusion. Diabetes Res Clin Pract 1994;25:1–12.ArticlePubMed
- 14. Moller L, Dalman L, Norrelund H, Billestrup N, Frystyk J, Moller N, et al. Impact of fasting on growth hormone signaling and action in muscle and fat. J Clin Endocrinol Metab 2009;94:965–72.ArticlePubMed
- 15. Blaine E, Tumlinson R, Colvin M, Haynes T, Whitley HP. Systematic literature review of insulin dose adjustments when initiating hemodialysis or peritoneal dialysis. Pharmacotherapy 2022;42:177–87.ArticlePubMedPDF
- 16. Agius R, Pace NP, Fava S. Phenotyping obesity: a focus on metabolically healthy obesity and metabolically unhealthy normal weight. Diabetes Metab Res Rev 2023;e3725.ArticlePubMed
- 17. Boutari C, DeMarsilis A, Mantzoros CS. Obesity and diabetes. Diabetes Res Clin Pract 2023;202:110773.ArticlePubMed
- 18. Savastano S, Di Somma C, Barrea L, Colao A. The complex relationship between obesity and the somatropic axis: the long and winding road. Growth Horm IGF Res 2014;24:221–6.ArticlePubMed
- 19. Nam SY, Kim KR, Cha BS, Song YD, Lim SK, Lee HC, et al. Low-dose growth hormone treatment combined with diet restriction decreases insulin resistance by reducing visceral fat and increasing muscle mass in obese type 2 diabetic patients. Int J Obes Relat Metab Disord 2001;25:1101–7.ArticlePubMedPDF
- 20. Ahn CW, Kim CS, Nam JH, Kim HJ, Nam JS, Park JS, et al. Effects of growth hormone on insulin resistance and atherosclerotic risk factors in obese type 2 diabetic patients with poor glycaemic control. Clin Endocrinol (Oxf) 2006;64:444–9.ArticlePubMed
- 21. Turner RC, Holman RR. Lessons from UK prospective diabetes study. Diabetes Res Clin Pract 1995;28 Suppl:S151–7.ArticlePubMed
- 22. McFarlane SI. Antidiabetic medications and weight gain: implications for the practicing physician. Curr Diab Rep 2009;9:249–54.ArticlePubMedPDF
- 23. Marasco G, Dajti E, Ravaioli F, Brocchi S, Rossini B, Alemanni LV, et al. Clinical impact of sarcopenia assessment in patients with liver cirrhosis. Expert Rev Gastroenterol Hepatol 2021;15:377–88.ArticlePubMed
- 24. Yen FS, Hou MC, Liu JS, Hsu CC, Hwu CM. Severe hypoglycemia in patients with liver cirrhosis and type 2 diabetes. Front Med (Lausanne) 2023;9:962337.ArticlePubMedPMC
- 25. Assaad SN, Cunningham GR, Samaan NA. Abnormal growth hormone dynamics in chronic liver disease do not depend on severe parenchymal disease. Metabolism 1990;39:349–56.ArticlePubMed
- 26. Finn PF, Dice JF. Proteolytic and lipolytic responses to starvation. Nutrition 2006;22:830–44.ArticlePubMed
- 27. de la Garza RG, Morales-Garza LA, Martin-Estal I, Castilla-Cortazar I. Insulin-like growth factor-1 deficiency and cirrhosis establishment. J Clin Med Res 2017;9:233–47.ArticlePubMedPMC
- 28. Melmed S. Acromegaly pathogenesis and treatment. J Clin Invest 2009;119:3189–202.ArticlePubMedPMC
- 29. Melmed S, Colao A, Barkan A, Molitch M, Grossman AB, Kleinberg D, et al. Guidelines for acromegaly management: an update. J Clin Endocrinol Metab 2009;94:1509–17.ArticlePubMed
- 30. Trainer PJ, Drake WM, Katznelson L, Freda PU, Herman-Bonert V, van der Lely AJ, et al. Treatment of acromegaly with the growth hormone–receptor antagonist pegvisomant. New Engl J Med 2000;342:1171–7.PubMed
- 31. van der Lely AJ, Hutson RK, Trainer PJ, Besser GM, Barkan AL, Katznelson L, et al. Long-term treatment of acromegaly with pegvisomant, a growth hormone receptor antagonist. Lancet 2001;358:1754–9.ArticlePubMed
- 32. Castinetti F, Nagai M, Morange I, Dufour H, Caron P, Chanson P, et al. Long-term results of stereotactic radiosurgery in secretory pituitary adenomas. J Clin Endocrinol Metab 2009;94:3400–7.ArticlePubMedPDF
- 33. Moller N, Jorgensen JO. Effects of growth hormone on glucose, lipid, and protein metabolism in human subjects. Endocr Rev 2009;30:152–77.ArticlePubMed
- 34. Jørgensen JO, Krag M, Jessen N, Norrelund H, Vestergaard ET, Moller N, et al. Growth hormone and glucose homeostasis. Horm Res 2004;62 Suppl 3:51–5.PubMed
- 35. Parkinson C, Flyvbjerg A, Trainer PJ. High levels of 150-kDa insulin-like growth factor binding protein three ternary complex in patients with acromegaly and the effect of pegvisomant-induced serum IGF-I normalization. Growth Horm IGF Res 2004;14:59–65.ArticlePubMed
- 36. Ho PJ, Friberg RD, Barkan AL. Regulation of pulsatile growth hormone secretion by fasting in normal subjects and patients with acromegaly. J Clin Endocrinol Metab 1992;75:812–9.ArticlePubMed
- 37. Coopmans EC, Berk KA, El-Sayed N, Neggers SJ, van der Lely AJ. Eucaloric very-low-carbohydrate ketogenic diet in acromegaly treatment. N Engl J Med 2020;382:2161–2.ArticlePubMed
- 38. Jorgensen JO, Rubeck KZ, Nielsen TS, Clasen BF, Vendelboe M, Hafstrom TK, et al. Effects of GH in human muscle and fat. Pediatr Nephrol 2010;25:705–9.ArticlePubMedPDF
References
Figure & Data
References
Citations
Citations to this article as recorded by 
- IGF-1 and IGF-2 as Molecules Linked to Causes and Consequences of Obesity from Fetal Life to Adulthood: A Systematic Review
Justyna Szydlowska-Gladysz, Adrianna Edyta Gorecka, Julia Stepien, Izabela Rysz, Iwona Ben-Skowronek
International Journal of Molecular Sciences.2024; 25(7): 3966. CrossRef
 PubReader
PubReader ePub Link
ePub Link Cite
Cite