State-of-the-Art Overview of the Pharmacological Treatment of Non-Alcoholic Steatohepatitis
Article information

Abstract
Non-alcoholic fatty liver disease (NAFLD) is the most common cause of chronic liver disease worldwide, and non-alcoholic steatohepatitis (NASH), a subtype of NAFLD, can progress to cirrhosis, hepatocellular carcinoma, and death. Nevertheless, the current treatment for NAFLD/NASH is limited to lifestyle modifications, and no drugs are currently officially approved as treatments for NASH. Many global pharmaceutical companies are pursuing the development of medications for the treatment of NASH, and results from phase 2 and 3 clinical trials have been published in recent years. Here, we review data from these recent clinical trials and reports on the efficacy of newly developed antidiabetic drugs in NASH treatment.
INTRODUCTION
Non-alcoholic fatty liver disease (NAFLD) is considered a liver manifestation of ectopic fat accumulation that is primarily caused by abdominal obesity and insulin resistance. Dysfunctional adipose tissue and increased lipolysis contribute to the development of insulin resistance and lipotoxicity in multiple organs [1]. Large amounts of free fatty acids (FFAs) from the visceral fat and dietary lipids enter the liver, and NAFLD is associated with decreased lipid excretion, increased accumulation of toxic lipids such as diacylglycerol, which induces hepatic insulin resistance, and oxidative stress. These can activate inflammatory and fibrogenic responses in multiple immune cells and hepatic stellate cells, leading to the progression of nonalcoholic steatohepatitis (NASH) [2,3].
The prevalence of NAFLD has increased concomitantly with increasing obesity rates. The incidence of NAFLD in the general population is about 25%, but increases to more than 90% in highly obese populations [3,4]. NAFLD is the most common cause of chronic liver disease in the world and NASH, a subtype of NAFLD, can progress to cirrhosis, hepatocellular carcinoma, and death [4]. Recent evidence also suggests that NASH should be considered an independent cardiovascular risk factor [5].
Nonetheless, the current treatment of NAFLD or NASH is limited to lifestyle modifications, as no drugs have been officially approved by the U.S. Food and Drug Administration (FDA) for NASH treatment. In several NAFLD treatment guidelines, all drugs currently used in NAFLD treatment are considered off-label treatments [6]. Vitamin E (an antioxidant) and pioglitazone (an antidiabetic agent) have been reported to be effective in improving NASH in randomized clinical trials, including the PIVENS trial [7]. However, the long-term safety of vitamin E is controversial due to its potential risk for increased mortality [8], and pioglitazone can increase body weight and fluid retention. Therefore, many global pharmaceutical companies are pursuing the development of medications for NASH treatment, and the results of phase 2 and 3 clinical trials have been published in recent years. Here, we review these recent clinical trial data and reports of the efficacy of newly developed antidiabetic drugs on NASH. A summary of recent clinical data is provided in Table 1.
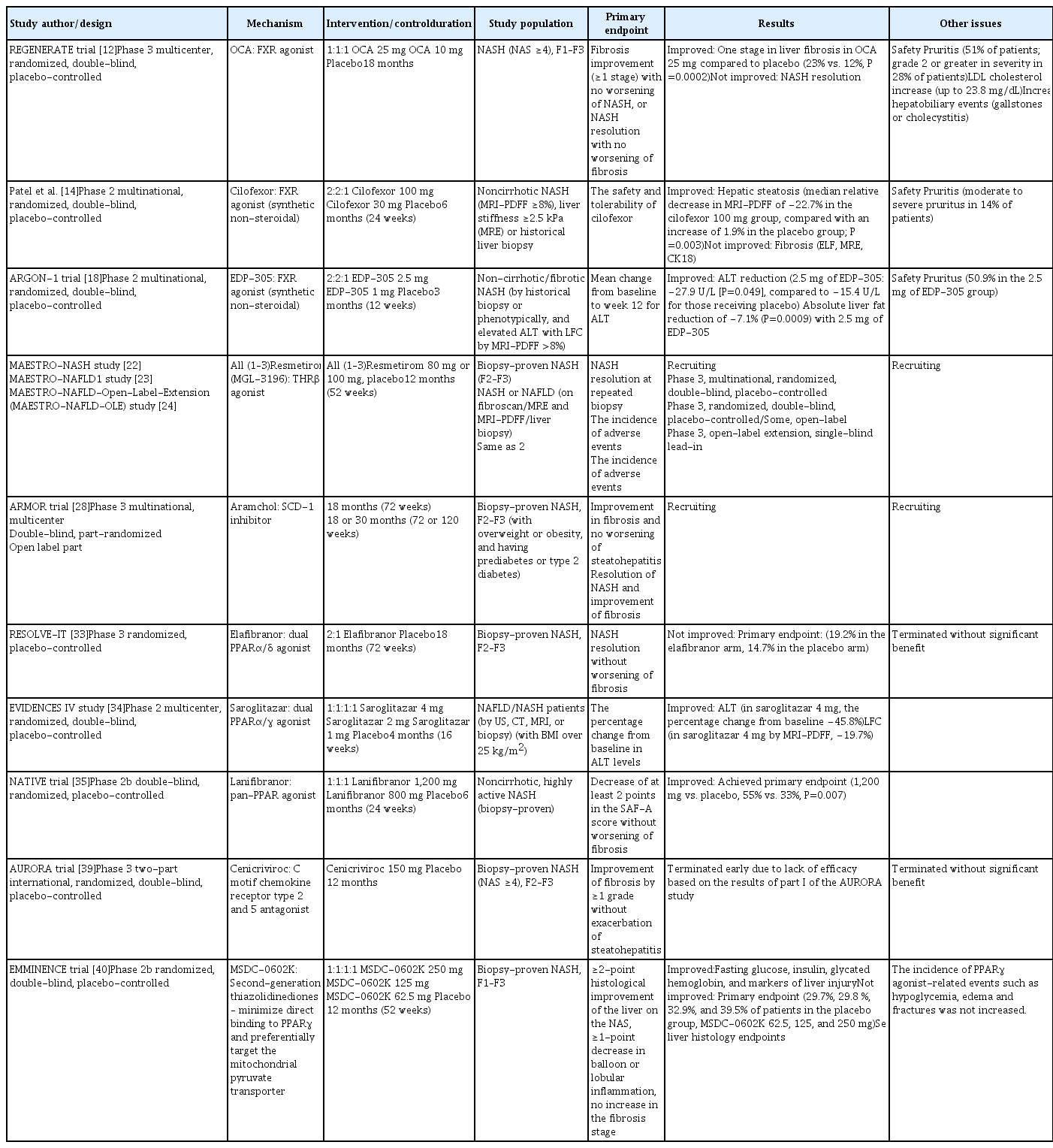
Recent Clinical Trial Data of NASH Therapeutics
TARGETING BILE ACID RECEPTORS/FARNESOID X RECEPTOR
Bile acids are natural ligands of the farnesoid X receptor (FXR) [9]. Together they regulate lipid and/or glucose homeostasis, promote insulin sensitivity, and potentially regulate liver fibrosis [10]. FXR agonists have been shown to prevent the development of NASH and promote the resolution of NASH and fibrosis in rodent models [11]. Multiple FXR agonists are now being tested for NASH treatment.
Obeticholic acid
The phase 3 REGENERATE trial (NCT02548351) evaluated histological response after 18 months of maintenance in 1,968 patients who received obeticholic acid (OCA; 10 or 25 mg) or placebo. The results demonstrated significantly more frequent improvement by ≥1 stage in liver fibrosis among patients receiving 25 mg of OCA daily compared to patients receiving placebo (23% vs. 12%, P=0.0002) [12]. However, treatment with OCA also raised concerns about several adverse events. OCA at a dose of 25 mg/day was associated with pruritus in 51% of patients. An increase in low-density lipoprotein (LDL) cholesterol levels of up to 23.8 mg/dL was observed after 1 month of OCA treatment at 25 mg/day, which led to statin treatment in a large number of patients. More patients (3%) who received 25 mg of OCA experienced hepatobiliary events (gallstones or cholecystitis) than those (<1%) who received placebo [12]. The FDA announced that the predicted benefit of OCA remains uncertain and does not sufficiently outweigh the potential risks to support accelerated approval for NASH treatment [13]. It will be necessary to confirm additional post-interim analysis efficacy and safety data from the ongoing REGENERATE study results.
Synthetic non-steroidal FXR agonists, including cilofexor [14], tropifexor [15], EDP-305 [16], and MET-409 [17] are in development, and phase 2 trials are ongoing. However, these compounds also show dose-dependent associations with pruritus. The similarity of their chemical structures to the steroid bile acid of OCA can contribute to the risk of the above drug adverse events.
Cilofexor
In a phase 2 trial, 140 patients with noncirrhotic NASH were randomized to receive cilofexor (30 or 100 mg) or placebo for 24 weeks [14]. Cilofexor was well-tolerated and provided significant reductions in hepatic steatosis (median relative decrease in magnetic resonance imaging proton density fat fraction [MRI-PDFF] of −22.7% in those receiving 100 mg of cilofexor compared with an increase of 1.9% in those receiving placebo) [14]. Cilofexor at a dose of 100 mg was associated with moderate to severe pruritus in 14% of patients (4% in the placebo group).
EDP-305
In an early phase 2 study of 134 non-cirrhotic patients with fibrotic NASH (ARGON-1), the primary endpoint was the mean change in alanine aminotransferase (ALT) levels from baseline to week 12, and the key secondary endpoint was the mean change in liver fat content (LFC) measured by MRI-PDFF. EDP-305 at a dose of 2.5 mg reduced ALT levels (–27.9 U/L, P=0.049) and liver fat (–7.1%, P=0.0009). However, pruritus occurred in 50.9% of patients in the 2.5 mg group [18].
TARGETING LIVER-SPECIFIC THYROID HORMONE RECEPTORS
Thyroid hormones are involved in the regulation of hepatic triglyceride and cholesterol metabolism [19]. Thyroid hormones reduce serum cholesterol by affecting cholesterol synthesis, LDL clearance, and reverse cholesterol transport. In this respect, thyroid hormone receptor-β (THR-β), which is mainly expressed in the liver, could be a target for NAFLD treatment [19]. Currently, resmetirom and VK2809 [20] are the THR agonists that mainly act on THR-β and are being developed for the treatment of NASH.
Resmetirom
When assessed by MRI-PDFF in a phase 2 trial, resmetirom-treated patients showed a relative reduction of hepatic fat compared with those who received placebo at week 36 (−37.3% for resmetirom vs. −8.5% for placebo, P<0.001) [21]. There was no significant difference in the proportion of patients with a ≥1-point reduction in fibrosis without worsening of the NAFLD activity score (NAS). Multiple atherogenic lipids and lipoproteins were also significantly reduced in patients who received resmetirom compared with placebo, particularly LDL cholesterol, apolipoprotein B, and triglycerides. The most common adverse effects were transient mild diarrhea and nausea [21]. There are three ongoing phase 3 trials with resmetirom [22–24]. The A Phase 3 Study to Evaluate the Efficacy and Safety of MGL-3196 (Resmetirom) in Patients With NASH and Fibrosis (MAESTRO-NASH) study is focusing on patients with biopsy-proven NASH (fibrosis stages 2 and 3 [F2–F3]), and its primary endpoint is NASH resolution at a repeated biopsy after 52 weeks of treatment [22].
TARGETING DE NOVO LIPOGENESIS
Increased de novo lipogenesis is one of the distinguishing features of NAFLD. This indicates that lipogenesis can be a therapeutic target for NAFLD.
Aramchol
Arachidyl amido cholanoic acid (Aramchol) downregulates stearoyl-CoA desaturase 1 (SCD-1), a key enzyme involved in hepatic lipogenesis [25]. By inhibiting SCD-1 activity, Aramchol downregulated the production of hepatic fatty acids and reduced fibrosis in mice [26]. The ARREST trial is a phase 2b trial evaluating the use of Aramchol (400 and 600 mg) in NASH patients with a body mass index of 25 to 40 kg/m2, corresponding to overweight or obesity, as well as prediabetes or type 2 diabetes mellitus (T2DM). Improvements in liver fat measured by magnetic resonance spectroscopy (MRS) were only significant in the group receiving 400 mg of Aramchol compared to placebo; however, the arm that received 600 mg of Aramchol had higher rates of NASH resolution. Decreases in ALT, aspartate aminotransferase, and glycated hemoglobin were also observed in the Aramchol group. The most common adverse drug reactions were urinary tract infection, headache, itching, and nausea [27]. The ARMOR trial (NCT04104321) is an ongoing phase 3/4 trial with NASH patients with fibrosis (F2–F3) to evaluate the safety and efficacy of Aramchol (600 mg). The primary endpoint is the proportion of patients with resolution of NASH without exacerbation of liver fibrosis and an improvement of stage 1 or greater in fibrosis without exacerbation of steatohepatitis [28].
Firsocostat (GS-0976)
Acetyl-coenzyme carboxylase catalyzes the rate-limiting step in de novo lipogenesis. In a phase 2 trial, the safety and efficacy of firsocostat, an inhibitor of acetyl-coenzyme A carboxylase in the liver, was evaluated. Twelve-week administration of firsocostat (20 mg) decreased hepatic steatosis, with a relative decrease of over 30% from baseline on MRI-PDFF occurring in 48% of patients (P=0.004 vs. placebo) and a greater median relative decrease (decrease of 29%) than those given placebo (decrease of 8%, P=0.002) [29]. However, in the treatment groups that received 20 or 5 mg of firsocostat, serum triglyceride levels increased by 11% and 13%, respectively, and 14% and 18% of patients, respectively, developed asymptomatic hypertriglyceridemia (>500 mg/dL).
TARGETING PEROXISOME PROLIFERATOR ACTIVATED RECEPTORS
Peroxisome proliferator activated receptor-α (PPARα) activity is associated with increased energy burning and reduced fat storage in the liver [30]. PPARα upregulates a number of genes that play roles in fatty acid oxidation and in phospholipid remodeling, and it inhibits hepatic inflammatory processes. Hepatic PPARβ/δ is involved in transforming potentially toxic lipids into less toxic molecules by regulating monounsaturated fatty acid synthesis and increasing AMP-activated protein kinase activity, thereby suppressing lipogenesis and glycogen synthesis, reducing gluconeogenesis, and increasing fatty acid oxidation. PPARβ/δ also stimulates anti-inflammatory responses in the liver [31].
Elafibranor
Elafibranor is a dual agonist of PPARα and PPARδ. In the phase 2 GOLDEN-505 trial, no statistically significant difference was observed in the primary outcome, the resolution of NASH without worsening of fibrosis [32]. In a post hoc analysis of patients with fibrosis (F2–F3), a higher rate of NASH resolution was seen in the group that received elafibranor (120 mg) daily than in the placebo group (19% vs. 12%, P=0.045). A phase 3 trial (RESOLVE-IT) compared the effects of treatment for 72 weeks in patients with histologically proven NASH with fibrosis (F2–F3). However, no statistically significant difference was found in the proportion of patients who experienced NASH resolution without worsening of fibrosis (19.2% in the elafibranor arm, 14.7% in the placebo arm) [33], which led to the termination of this study.
Saroglitazar
Saroglitazar is a dual PPARα/γ agonist. In the phase 2 EVIDENCES IV study (NCT03061721), 106 NAFLD/NASH patients were randomized to receive either placebo or saroglitazar (1, 2, or 4 mg). Compared with placebo, 4 mg of saroglitazar significantly improved ALT levels at 16 weeks (3.4% vs. −45.8%) and LFC as determined by MRI-PDFF (4.1% vs. −19.7%) [34].
Lanifibranor
Lanifibranor is a pan-PPAR agonist. In the NATIVE trial (NCT03008070), a total of 247, non-cirrhotic, highly active NASH patients were randomized to receive lanifibranor (800 or 1,200 mg) or placebo for 24 weeks. The primary endpoint was a decrease of at least 2 points in the activity part of the Steatosis, Activity, Fibrosis scoring system, which incorporates scores for ballooning and inflammation, without worsening of fibrosis. The percentage of patients who achieved the primary endpoint was significantly higher with the 1,200-mg dose of lanifibranor than with placebo (55% vs. 33%, P=0.007) [35].
TARGETING ANTI-INFLAMMATORY SIGNALING: C MOTIF CHEMOKINE RECEPTOR 2/5
Cenicriviroc
C motif chemokine receptor types 2 and 5 are both involved in the inflammatory and fibrogenic pathways [36]. Cenicriviroc binds to these chemokine receptors as a dual antagonist, and may reduce inflammation and improve NASH [37]. The CENTAUR phase 2 trial enrolled NASH patients with NAS ≥4 and liver fibrosis (stages 1–3, NASH clinical research network). The primary outcome, a ≥2-point improvement in NAS and no worsening of fibrosis at year 1, was achieved in similar proportions of subjects on cenicriviroc and placebo (P>0.05). However, the fibrosis endpoint (improvement in fibrosis by ≥1 stage) was met in more subjects with cenicriviroc than with placebo (20% vs. 10%, P=0.023) [38]. A subsequent phase 3 AURORA trial in patients with NASH and fibrosis (F2–F3) was conducted to evaluate the primary endpoint of improvement in fibrosis by ≥1 grade without exacerbation of steatohepatitis at 1 year. However, this study was terminated early due to lack of efficacy [39].
INSULIN SENSITIZER
MSDC-0602K
MSDC-0602K is a second-generation thiazolidinedione (TZD) designed to minimize direct binding to PPARγ and preferentially target the mitochondrial pyruvate transporter. In the EMMINENCE phase 2b trial (NCT0278444), patients with biopsy-confirmed NASH and fibrosis (F1–F3) were randomized to placebo or MSDC-0602K for 52 weeks. The primary endpoint was a ≥2-point histological improvement of the liver on the NAS, a ≥1 point decrease in balloon or lobular inflammation, and no increase in the fibrosis stage at 12 months [40]. MSDC-0602K significantly decreased fasting glucose, insulin, glycated hemoglobin, and markers of liver injury. However, the results for the primary endpoint and liver histology outcomes did not show statistically significant improvements in the MSDC-0602K group [40].
OTHER PHASE 2 CLINICAL TRIALS
Belapectin, a galectin 3 inhibitor (anti-fibrotic)
Increased levels of galectin 3 have been associated with NASH development. Belapectin, an inhibitor of galectin 3, reduced liver fibrosis in rats [41] and was well tolerated in phase 1 studies [42]. However, receiving biweekly infusions of belapectin for 1 year was not associated with a significant reduction in fibrosis compared with placebo in a phase 2b study conducted in 162 patients with NASH, cirrhosis, and portal hypertension [43].
Emricasan, a caspase inhibitor (apoptosis)
Lipotoxicity activates caspases that trigger apoptosis and the production of inflammatory cytokines. However, pan-caspase inhibition with emricasan did not improve liver histology, and may have worsened fibrosis and ballooning in patients with NASH and fibrosis (F1–F3) [44].
Aldafermin (NGM282), a fibroblast growth factor-19 analogue (hormone signaling)
Fibroblast growth factor 19 (FGF-19) plays a central role in regulating bile acids and energy metabolism in the liver via FGF receptor 4 [45]. Aldafermin, an analogue of FGF19, ameliorated hepatic steatosis with a reduction of 5.0% in absolute LFC (P=0.002) in a phase 2 trial of patients with NASH [46]. In the phase 2b ALPINE 2/3 clinical trial in NASH patients with fibrosis (F2–F3), aldafermin did not meet the primary endpoint of fibrosis improvement by ≥1 stage with no worsening of NASH versus placebo, and the trial was terminated [47]. A phase 2b ALPINE 4 study in NASH and fibrosis (F4) patients with compensated cirrhosis is still ongoing.
Pegbelfermin and efruxifermin, fibroblast growth factor-21 analogues (hormone signaling)
FGF-21 also plays a central role in regulating energy metabolism [45]. Pegbelfermin (BMS-986036) [48] and efruxifermin [49], which are FGF-21 analogues, significantly reduced the hepatic fat fraction in patients with NASH in phase 2 trials.
ANTIDIABETIC DRUGS FOR NAFLD MANAGEMENT
T2DM results from multiple organ abnormalities and various pathophysiological abnormalities. It has been linked to insulin resistance and obesity, indicating that T2DM and NAFLD have similar pathophysiological underpinnings [2,50]. The recently proposed term “metabolic (dysfunction)-associated fatty liver disease (MAFLD)” suggests the importance of an approach to NAFLD that recognizes the role of metabolic dysfunction in driving the pathophysiology [51]. Adipose tissue dysfunction and insulin resistance, which is the main feature of T2DM, increase the release of pro-inflammatory cytokines and decrease the release of anti-inflammatory adipokines [52]. These changes can directly damage the liver or act indirectly, leading to increased oxidative stress, hepatocyte damage, progression of hepatic fibrosis, and tumor development [53]. Glucotoxicity also promotes NASH and disease progression by stimulating the de novo synthesis of FFAs and ectopic fat accumulation [54]. This suggests that T2DM risk reduction and the improvement or resolution of NAFLD are closely related [55]. Most of the drugs presented below are used for the treatment of T2DM, and many clinical studies are being conducted on patients without T2DM. Therefore, there are relatively few areas of concern in terms of safety. A summary of newly developed antidiabetic medications is provided in Table 2.

Efficacy of Antidiabetic Drugs on NASH Treatment
ANTIDIABETIC DRUGS FOR NAFLD MANAGEMENT: THIAZOLIDINEDIONE
First-generation insulin-sensitizing TZDs bind directly to and activate the PPARγ nuclear hormone receptor [56]. The induction of PPARγ promotes the differentiation of large insulin-resistant pre-adipocytes into small, insulin-sensitive adipocytes [57]. This leads to an increase in FFA uptake in adipocytes, and the FFA burden is transferred to adipocytes rather than the liver [58]. TZDs have been shown to improve insulin resistance and glucose metabolism and are still used to treat T2DM.
Pioglitazone
TZD-based drugs have been reported to be effective in improving NAFLD in many human studies. In a pioglitazone trial reported in 2016 (NCT00994682), all patients (n=101) with prediabetes or T2DM and biopsy-proven NASH were prescribed a low-calorie diet and then randomly assigned to pioglitazone (45 mg/day) or placebo for 18 months. Of the patients with pioglitazone, 58% achieved the primary outcome (≥2 point decrease in NAS without worsening of fibrosis vs. 17% of the placebo group, P<0.001) and 51% had resolution of NASH (vs. 19% of the placebo group, P<0.001) [59].
ANTIDIABETIC DRUGS FOR NAFLD MANAGEMENT: GLUCAGON-LIKE PEPTIDE 1
Glucagon-like peptide 1 (GLP-1) analogues have been reported to be involved in weight loss and systemic insulin resistance reduction, which may lead to improvements in NAFLD. GLP-1 analogues may also act directly on human hepatocytes to decrease de novo adipogenesis and increase fatty acid oxidation, thereby ameliorating NAFLD [60].
Liraglutide
In the LEAN phase 2 trial, the effectiveness of subcutaneous injections of liraglutide (1.8 mg/day) versus placebo for 48 weeks in overweight patients with NASH was evaluated. Nine of 23 patients (39%) who received liraglutide showed resolution of NASH without worsening of fibrosis, compared to two of 22 patients (9%) in the placebo group (P=0.019) [61].
Semaglutide
In a 72-week phase 2 trial involving patients with biopsy-proven NASH and fibrosis (F1–F3, NCT02970942), 320 patients were randomly assigned, in a 3:3:3:1:1:1 ratio, to receive once-daily subcutaneous semaglutide at a dose of 0.1, 0.2, or 0.4 mg or the corresponding placebo. The primary endpoint (resolution of NASH with no worsening of fibrosis) was achieved in 40% of the 0.1 mg group, 36% of the 0.2 mg group, 59% of the 0.4 mg group, and 17% of the placebo group (P<0.001 for semaglutide [0.4 mg] vs. placebo). However, significant between-group differences in the improvement of the fibrosis stage were not observed. The mean percent weight loss was 13% in the 0.4 mg group (1% in the placebo group) [62].
Dulaglutide
Data regarding the effect of dulaglutide, a once-weekly GLP-1R agonist, on NAFLD are limited. The D-LIFT trial was a 24 week, open-label, randomized controlled trial (NCT03590626) conducted in India to determine the effect of dulaglutide (0.75 mg weekly for 4 weeks, followed by 1.5 mg weekly for 20 weeks) on liver fat in 64 patients who had T2DM with MRI-PDFF ≥6.0%. Dulaglutide treatment led to significant reductions in LFC, with a control-corrected absolute change in MRI-PDFF of −3.5% (P=0.025) and a relative change of −26.4% (P=0.004) [63].
ANTIDIABETIC DRUGS FOR NAFLD MANAGEMENT: SODIUM-GLUCOSE COTRANSPORTER 2 INHIBITOR
Sodium-glucose cotransporter 2 inhibitors (SGLT2i) are increasingly used antidiabetic drugs that increase urinary glucose excretion by inhibiting the reabsorption of glucose in the proximal tubule of the kidney [64]. Inhibition of SGLT2 causes an additional 60 to 80 g of glucose per day to be excreted out of the body [65], which leads to caloric loss and weight reduction [66]. There is a growing expectation that this mechanism of SGLT2i will assist in the improvement of NAFLD.
Dapagliflozin
In the EFFECTII study (NCT02279407), which aimed to investigate the effects of dapagliflozin and omega-3 (n-3) carboxylic acids, 84 participants with T2DM and NAFLD were randomly assigned 1:1:1:1 to receive 10 mg of dapagliflozin, 4 g of omega-3 (n-3) carboxylic acids, a combination of both, or placebo. After 12 weeks, only the combination treatment significantly reduced LFC as determined by MRI-PDFF (P=0.046) and total liver fat volume (relative change −24%, P=0.037) in comparison with placebo [67]. In 32 obese patients with T2DM, 8 weeks of treatment with dapagliflozin significantly reduced LFC as determined by MRI-PDFF (placebo-corrected decrease −3.74%, P<0.01) [68].
Empagliflozin
In the E-LIFT trial (50 patients with T2DM and NAFLD defined by MRI-PDFF >6%), 10 mg of empagliflozin led to a significant reduction in liver fat at 20 weeks (MRI-PDFF difference between groups −4.0%, P<0.001) [69]. In another randomized, double-blind, phase 4 trial with T2DM patients (n= 84), 25 mg of empagliflozin treatment resulted in a placebo-corrected absolute change of −1.8% (P=0.02) in LFC as determined by MRS and a relative change of −22% (P=0.009) at 24 weeks [70].
Canagliflozin
In a double-blind, 24-week trial in 56 subjects with inadequately controlled T2DM (NCT02009488), changes in intrahepatic triglyceride content (IHTG) measured by MRS, insulin sensitivity, and beta-cell function were compared after treatment with canagliflozin (300 mg) or placebo for 6 months [71]. Although canagliflozin significantly improved hepatic insulin sensitivity, only a numerically larger absolute decrease in IHTG was observed (−4.6% vs. placebo −2.4%, P=0.09). In patients with NAFLD (n=37), the decrease in IHTG was −6.9 % vs. −3.8 % (P=0.05).
In summary, SGLT2i agents have the potential to improve NAFLD with their specific effect on weight reduction; however, the evidence for histological improvement and benefits in NAFLD patients without T2DM remains unclear.
COMBINATION APPROACHES
Since NAFLD results from multiple cellular and molecular disturbances occurring in multiple organ systems, efforts to increase the therapeutic effect by simultaneously targeting multiple mechanisms rather than a single mechanism have been proposed [72]. There are clinical studies with combinations of new drugs [73], and a number of clinical studies on GLP-1 based dual or triple agonists are in progress [74].
Tirzepatide
Tirzepatide, a dual agonist of the gastric inhibitory polypeptide (GIP) receptor and GLP-1 receptor, has been shown to lead to significant reductions in body weight and improvement of glycemic control in patients with T2DM [75–77]. Based on these studies, a study to confirm the therapeutic potential for non-cirrhotic NASH patients is in progress (A Study of Tirzepatide (LY3298176) in Participants With Nonalcoholic Steatohepatitis [SYNERGY-NASH], NCT04166773). This phase 2 trial comparing the efficacy and safety of tirzepatide versus placebo in patients with NASH and fibrosis (F2–F3) is recruiting participants with study completion planned for 2023. The primary endpoint is the percentage of participants with the absence of NASH and no worsening of fibrosis on liver histology at week 52 [78].
Cotadutide
Cotadutide, a dual GLP-1 and glucagon receptor agonist, is also under development for NASH treatment. In a recent trial, cotadutide treatment for 54 weeks improved glycemic control and weight loss in patients with overweight/obesity and T2DM [79]. Thus, cotadutide can be a promising therapeutic option for the treatment of NASH via modulating mitochondrial function and lipogenesis [80]. A recent phase 2 trial was conducted to evaluate the safety (including hepatic safety), tolerability and pharmacodynamic effects in 74 participants with biopsy-confirmed NAFLD/NASH with fibrosis (F1–F3) (NCT04019561, completed/not published) [81].
CONCLUSIONS
We reviewed recent clinical trial data of newly developed drugs for NASH treatment and reports of the efficacy of antidiabetic drugs on NASH or NAFLD. Although we should wait for final results of many ongoing clinical trials, the recent failure of NASH trials strongly suggests that simply targeting one pathway or mechanism is not sufficient to ameliorate NASH. This underscores the need to find the common root of NASH progression and candidates that can modulate multiple pathways or disease cascades in NASH pathophysiology [74]. In that respect, emerging multi-target therapies or combinations of antidiabetic agents with proven clinical efficacy and safety may also be promising options for the treatment of NASH, and more clinical investigations should be conducted in the future.
Acknowledgements
This work was supported by a research grant from the Inha University to Yongin Cho and Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2018R1D1A1B07050005) and the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (HI14C1324, HR18C0012020019) to Yong-ho Lee.
Notes
CONFLICTS OF INTEREST
No potential conflict of interest relevant to this article was reported.