
Search
- Page Path
- HOME > Search
Original Articles
- Thyroid
- Whole-Exome Sequencing in Papillary Microcarcinoma: Potential Early Biomarkers of Lateral Lymph Node Metastasis
- Mijin Kim, Chae Hwa Kwon, Min Hee Jang, Jeong Mi Kim, Eun Heui Kim, Yun Kyung Jeon, Sang Soo Kim, Kyung-Un Choi, In Joo Kim, Meeyoung Park, Bo Hyun Kim
- Endocrinol Metab. 2021;36(5):1086-1094. Published online October 28, 2021
- DOI: https://doi.org/10.3803/EnM.2021.1132

- 3,549 View
- 108 Download
- 4 Web of Science
- 4 Crossref
-
 Abstract
Abstract
 PDF
PDF Supplementary Material
Supplementary Material PubReader
PubReader  ePub
ePub - Background
Early identification of patients with high-risk papillary thyroid microcarcinoma (PTMC) that is likely to progress has become a critical challenge. We aimed to identify somatic mutations associated with lateral neck lymph node (LN) metastasis (N1b) in patients with PTMC.
Methods
Whole-exome sequencing (WES) of 14 PTMCs with no LN metastasis (N0) and 13 N1b PTMCs was performed using primary tumors and matched normal thyroid tissues.
Results
The mutational burden was comparable in N0 and N1b tumors, as the median number of mutations was 23 (range, 12 to 46) in N0 and 24 (range, 12 to 50) in N1b PTMC (P=0.918). The most frequent mutations were detected in PGS1, SLC4A8, DAAM2, and HELZ in N1b PTMCs alone, and the K158Q mutation in PGS1 (four patients, Fisher’s exact test P=0.041) was significantly enriched in N1b PTMCs. Based on pathway analysis, somatic mutations belonging to the receptor tyrosine kinase-RAS and NOTCH pathways were most frequently affected in N1b PTMCs. We identified four mutations that are predicted to be pathogenic in four genes based on Clinvar and Combined Annotation-Dependent Depletion score: BRAF, USH2A, CFTR, and PHIP. A missense mutation in CFTR and a nonsense mutation in PHIP were detected in N1b PTMCs only, although in one case each. BRAF mutation was detected in both N0 and N1b PTMCs.
Conclusion
This first comprehensive WES analysis of the mutational landscape of N0 and N1b PTMCs identified pathogenic genes that affect biological functions associated with the aggressive phenotype of PTMC. -
Citations
Citations to this article as recorded by
- Feasibility of whole‐exome sequencing in fine‐needle aspiration specimens of papillary thyroid microcarcinoma for the identification of novel gene mutations
Liyuan Ma, Luying Gao, Ya Hu, Xiaoyi Li, Chunhao Liu, Jiang Ji, Xinlong Shi, Aonan Pan, Yuang An, Nengwen Luo, Yu Xia, Yuxin Jiang
Clinical Genetics.2024; 105(5): 567. CrossRef - Multi-omics analysis reveals a molecular landscape of the early recurrence and early metastasis in pan-cancer
Dan-ni He, Na Wang, Xiao-Ling Wen, Xu-Hua Li, Yu Guo, Shu-heng Fu, Fei-fan Xiong, Zhe-yu Wu, Xu Zhu, Xiao-ling Gao, Zhen-zhen Wang, Hong-jiu Wang
Frontiers in Genetics.2023;[Epub] CrossRef - What can we learn about acid-base transporters in cancer from studying somatic mutations in their genes?
Bobby White, Pawel Swietach
Pflügers Archiv - European Journal of Physiology.2023;[Epub] CrossRef - Comprehensive Long-Read Sequencing Analysis Discloses the Transcriptome Features of Papillary Thyroid Microcarcinoma
Yanqiang Wang, Binbin Zou, Yanyan Zhang, Jin Zhang, Shujing Li, Bo Yu, Zhekun An, Lei Li, Siqian Cui, Yutong Zhang, Jiali Yao, Xiuzhi Shi, Jing Liu
The Journal of Clinical Endocrinology & Metabolism.2023;[Epub] CrossRef
- Feasibility of whole‐exome sequencing in fine‐needle aspiration specimens of papillary thyroid microcarcinoma for the identification of novel gene mutations

- Clinical Study
- Whole Exome Sequencing Identifies Novel Genetic Alterations in Patients with Pheochromocytoma/Paraganglioma
- Soo Hyun Seo, Jung Hee Kim, Man Jin Kim, Sung Im Cho, Su Jin Kim, Hyein Kang, Chan Soo Shin, Sung Sup Park, Kyu Eun Lee, Moon-Woo Seong
- Endocrinol Metab. 2020;35(4):909-917. Published online December 23, 2020
- DOI: https://doi.org/10.3803/EnM.2020.756
- 5,190 View
- 154 Download
- 10 Web of Science
- 8 Crossref
-
Abstract
PDFSupplementary MaterialPubReader ePub
- Background
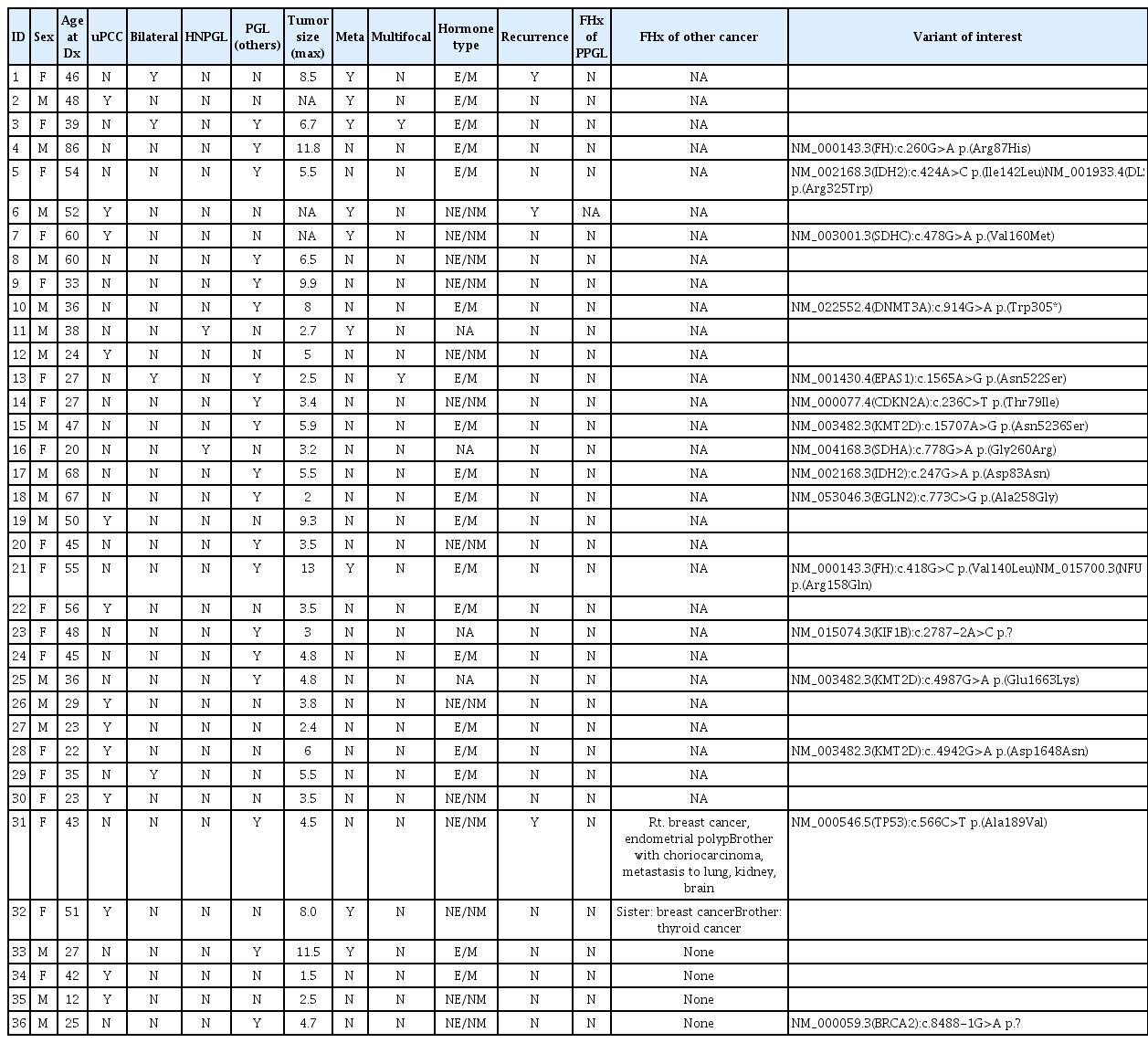
Pheochromocytoma and paragangliomas (PPGL) are known as tumors with the highest level of heritability, approximately 30% of all cases. Clinical practice guidelines of PPGL recommend genetic testing for germline variants in all patients. In this study, we used whole exome sequencing to identify novel causative variants associated with PPGL to improve the detection of rare genetic variants in our cohort.
Methods
Thirty-six tested negative for pathogenic variants in previous Sanger sequencing or targeted gene panel testing for PPGL underwent whole exome sequencing. Whole exome sequencing was performed using DNA samples enriched using TruSeq Custom Enrichment Kit and sequenced with MiSeq (Illumina Inc.). Sequencing alignment and variant calling were performed using SAMtools.
Results
Among previously mutation undetected 36 patients, two likely pathogenic variants and 13 variants of uncertain significance (VUS) were detected in 32 pheochromocytoma-related genes. SDHA c.778G>A (p.Gly260Arg) was detected in a patient with head and neck paraganglioma, and KIF1B c.2787-2A>C in a patient with a bladder paraganglioma. Additionally, a likely pathogenic variant in BRCA2, VUS in TP53, and VUS in NFU1 were detected.
Conclusion
Exome sequencing further identified genetic alterations by 5.6% in previously mutation undetected patients in PPGL. Implementation of targeted gene sequencing consisted of extended genes of PPGL in routine clinical screening can support the level of comprehensive patient assessment. -
Citations
Citations to this article as recorded by- Patient Sex and Origin Influence Distribution of Driver Genes and Clinical Presentation of Paraganglioma
Susan Richter, Nicole Bechmann
Journal of the Endocrine Society.2024;[Epub] CrossRef - Case Report: Aggressive neural crest tumors in a child with familial von Hippel Lindau syndrome associated with a germline VHL mutation (c.414A>G) and a novel KIF1B gene mutation
Lucie Landen, Anne De Leener, Manon Le Roux, Bénédicte Brichard, Selda Aydin, Dominique Maiter, Philippe A. Lysy
Frontiers in Endocrinology.2023;[Epub] CrossRef - A Case of Pheochromocytoma as a Subsequent Neoplasm in a Survivor of Childhood Embryonal Rhabdomyosarcoma
Rozalyn L. Rodwin, Sanyukta K. Janardan, Erin W. Hofstatter, Nina S. Kadan-Lottick
Journal of Pediatric Hematology/Oncology.2022; 44(2): e585. CrossRef - Mutations of 1p genes do not consistently abrogate tumor suppressor functions in 1p-intact neuroblastoma
Chik Hong Kuick, Jia Ying Tan, Deborah Jasmine, Tohari Sumanty, Alvin Y. J. Ng, Byrrappa Venkatesh, Huiyi Chen, Eva Loh, Sudhanshi Jain, Wan Yi Seow, Eileen H. Q. Ng, Derrick W. Q. Lian, Shui Yen Soh, Kenneth T. E. Chang, Zhi Xiong Chen, Amos H. P. Loh
BMC Cancer.2022;[Epub] CrossRef - A case of juvenile-onset pheochromocytoma with KIF1B p.V1529M germline mutation
Masahiro Nezu, Yosuke Hirotsu, Kenji Amemiya, Miho Katsumata, Tomomi Watanabe, Soichi Takizawa, Masaharu Inoue, Hitoshi Mochizuki, Kyoko Hosaka, Toshio Oyama, Masao Omata
Endocrine Journal.2022; 69(6): 705. CrossRef - Systematic analysis of expression profiles and prognostic significance for MMDS-related iron–sulfur proteins in renal clear cell carcinoma
Ling Yang, Yu-Xin Chen, Ying-Ying Li, Xiao-Juan Liu, Yong-Mei Jiang, Jia Mai
Scientific Reports.2022;[Epub] CrossRef - Diagnosis for Pheochromocytoma and Paraganglioma: A Joint Position Statement of the Korean Pheochromocytoma and Paraganglioma Task Force
Eu Jeong Ku, Kyoung Jin Kim, Jung Hee Kim, Mi Kyung Kim, Chang Ho Ahn, Kyung Ae Lee, Seung Hun Lee, You-Bin Lee, Kyeong Hye Park, Yun Mi Choi, Namki Hong, A Ram Hong, Sang-Wook Kang, Byung Kwan Park, Moon-Woo Seong, Myungshin Kim, Kyeong Cheon Jung, Chan
Endocrinology and Metabolism.2021; 36(2): 322. CrossRef - Recurrent Germline Mutations of CHEK2 as a New Susceptibility Gene in Patients with Pheochromocytomas and Paragangliomas
Yinjie Gao, Chao Ling, Xiaosen Ma, Huiping Wang, Yunying Cui, Min Nie, Anli Tong, Dario De Biase
International Journal of Endocrinology.2021; 2021: 1. CrossRef
- Patient Sex and Origin Influence Distribution of Driver Genes and Clinical Presentation of Paraganglioma
- Clinical Study
- Genetic Analysis of CLCN7 in an Old Female Patient with Type II Autosomal Dominant Osteopetrosis
- Seon Young Kim, Younghak Lee, Yea Eun Kang, Ji Min Kim, Kyong Hye Joung, Ju Hee Lee, Koon Soon Kim, Hyun Jin Kim, Bon Jeong Ku, Minho Shong, Hyon-Seung Yi
- Endocrinol Metab. 2018;33(3):380-386. Published online September 18, 2018
- DOI: https://doi.org/10.3803/EnM.2018.33.3.380
- 4,101 View
- 57 Download
- 2 Web of Science
- 2 Crossref
-
Abstract
PDFPubReader ePub
Background Type II autosomal dominant osteopetrosis (ADO II) is a rare genetically heterogeneous disorder characterized by osteosclerosis and increased bone mass, predominantly involving spine, pelvis, and skull. It is closely related to functional defect of osteoclasts caused by chloride voltage-gated channel 7 (
CLCN7 ) gene mutations. In this study, we aimed to identify the pathogenic mutation in a Korean patient with ADO II using whole exome sequencing.Methods We evaluated the clinical, biochemical, and radiographic analysis of a 68-year-old woman with ADO II. We also performed whole exome sequencing to identify pathogenic mutation of a rare genetic disorder of the skeleton. Moreover, a polymorphism phenotyping program, Polymorphism Phenotyping v2 (PolyPhen-2), was used to assess the effect of the identified mutation on protein function.
Results Whole exome sequencing using peripheral leukocytes revealed a heterozygous c.296A>G missense mutation in the
CLCN7 gene. The mutation was also confirmed using Sanger sequencing. The mutation c.296A>G was regarded to have a pathogenic effect by PolyPhen-2 software.Conclusion We detect a heterozygous mutation in
CLCN7 gene of a patient with ADO II, which is the first report in Korea. Our present findings suggest that symptoms and signs of ADO II patient having a c.296A>G mutation inCLCN7 may appear at a very late age. The present study would also enrich the database ofCLCN7 mutations and improve our understanding of ADO II.-
Citations
Citations to this article as recorded by- Autosomal dominant osteopetrosis type II resulting from a de novo mutation in the CLCN7 gene: A case report
Xiu-Li Song, Li-Yuan Peng, Dao-Wen Wang, Hong Wang
World Journal of Clinical Cases.2022; 10(20): 6936. CrossRef - Magnetic resonance findings in a Cavalier King Charles spaniel with osteopetrosis, Chiari‐like malformation and syringomyelia
Ricardo Fernandes, C J Jordan, Colin Driver
Veterinary Record Case Reports.2019;[Epub] CrossRef
- Autosomal dominant osteopetrosis type II resulting from a de novo mutation in the CLCN7 gene: A case report
 First
First Prev
Prev


