Articles
- Page Path
- HOME > Endocrinol Metab > Volume 28(3); 2013 > Article
-
Case ReportPolyglandular Autoimmune Syndrome Type III with Primary Hypoparathyroidism
- Sang Jin Kim1, Sang-Yoon Kim1, Han-Byul Kim1, Hyukwon Chang2, Ho-Chan Cho1
-
Endocrinology and Metabolism 2013;28(3):236-240.
DOI: https://doi.org/10.3803/EnM.2013.28.3.236
Published online: September 13, 2013
1Department of Internal Medicine, Keimyung University School of Medicine, Daegu, Korea.
2Department of Radiology, Keimyung University School of Medicine, Daegu, Korea.
- Corresponding author: Ho-Chan Cho. Department of Internal Medicine, Keimyung University School of Medicine, 56 Dalseong-ro, Jung-gu, Daegu 700-712, Korea. Tel: +82-53-250-7951, Fax: +82-53-250-7982, ho3632@naver.com
• Received: August 22, 2012 • Accepted: January 4, 2013
Copyright © 2013 Korean Endocrine Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- 3,927 Views
- 34 Download
- 5 Crossref
ABSTRACT
- Polyglandular autoimmune syndrome is defined as multiple endocrine gland insufficiencies accompanied by autoimmune diseases of the endocrine and nonendocrine system. After Schmidt introduced a case of nontuberculosis adrenal gland dysfunction with thyroiditis in 1926, Neufeld defined polyglandular autoimmune syndrome by I, II, and III subtypes in 1980 by their presentation of occurrence age, heredity methods, relationship with human leukocyte antigen, and accompanying diseases. We report a case of a 32-year-old female with polyglandular autoimmune syndrome III accompanied by type 1 diabetes mellitus that was treated with insulin (36 units per day) for 11 years. She had insulin deficiency and Hashimoto thyroiditis as an autoimmune disorder. In addition, she had several features similar to Albright's hereditary osteodystrophy including short stature, truncal obesity, round face, short neck, low intelligence (full IQ 84), and decreased memory. Although Albright's hereditary osteodystrophy is morphological evidence of pseudohypoparathyroidism or pseudopseudohypoparathyroidism, she had primary hypoparathyroidism on laboratory results. Here, we report a case of polyglandular autoimmune syndrome III with type 1 diabetes mellitus, autoimmune thyroiditis, and primary hypoparathyroidism, accompanied by clinical features similar to Albright's hereditary osteodystrophy.
- Polyglandular autoimmune syndrome (PAS) is defined as the functional disorder of two abnormal endocrine glands. In 1926, Schmidt reported a case in which nontuberculosis hypoadrenalism with autoimmune thyroiditis, and Neufeld et al. [1] divided the PAS into type I, II, and III according to the endocrinopathy occurrence age, heredity method, relationship with human leukocyte antigen (HLA), and the characteristics of accompanying disease. In 1980 [1], PAS type I corresponds to cases with at least two of the following: autoimmune Addison's disease, chronic mucocutaneous candidiasis, and idiopathic hypoparathyroidism, while PAS type II was defined as a disease group including Addison's disease, autoimmune thyroid disease, and type 1 diabetes [2]. PAS type III is not accompanied by Addison disease but includes other autoimmune diseases, such as type 1 diabetes together with autoimmune thyroid disease. In addition, Neufeld et al. [1] subdivided cases with both type 1 diabetes and autoimmune thyroiditis into PAS type IIIa. However, type I PAS is so rare that only 100 cases have been reported worldwide [3]. In Korea, some PAS cases with various clinical features including PAS III have been reported [4-8].
- Albright's hereditary osteodystrophy (AHO) is a growth impairment-related congenital disease, accompanied by several pathognomonic features [9,10]. These somatic features are associated with resistance to parathyroid hormone (PTH) including pseudohypoparathyroidism (PHP) I or pseudopseudohypoparathyroidism (PPHP) in an autosomal dominant inheritance [11]. Primary hypoparathyroidism is characterized as a deficiency or secretory disorder of PTH without underlying disorders including any prior neck surgery, chronic renal failure, alcoholism, gastrointestinal absorption disorders, or pathognomonic musculoskeletal abnormality such as AHO [12].
- In the present case, the 32-year-old female with type I diabetes had autoimmune thyroiditis, and therefore we could diagnosed her illness as having PAS type IIIa. At the same time, the present case was accompanied by primary hypoparathyroidism with clinical features similar to AHO and thyroid papillary cancer, and this is a rare case. Therefore, we report this valuable experience.
INTRODUCTION
- Patient
- 32-year-old female.
- Chief complaint
- General weakness.
- Present illness
- She was receiving insulin injections for type 1 diabetes, but her symptoms had not been improved at all, which prompted her decision to come to our hospital.
- Past history
- She had been using 32 U insulin every day for type 1 diabetes for 11 years before her visit to our hospital.
- Family history
- All family members, including both parents and all siblings, had diabetes.
- Physical examination
- When she appeared, her blood pressure was 130/80 mm Hg, pulse rate was 74 beats per minute, and body temperature was 36.6℃. She had no other symptoms, except for general weakness and central obesity (body mass index 23.4 kg/m2). No tumor-like mass was checked at her head and neck, nothing was discovered in chest auscultation. When we checked her abdomen, neither any pain nor hepatosplenomegaly was observed. No skin or oral mucosa pigmentation was found. There was no edema in lower leg. The neurological examination results were also normal. She weighed 51.5 kg and was 148.2 cm tall, and she was somewhat short woman with a short neck and round face, similar to a clinical features of AHO, but there was no pathognomonic brachydactyly.
- Laboratory results
- The result of complete blood count was the hemoglobin 9.3 g/dL, the erythrocyte average volume 96 fL, leukocyte 5,720/mm3, and platelets 310,000/mm. The results of anemia work up were the total iron binding capacity 447.5 µg/dL, serum ferritin 35.11 ng/mL, reticulocyte count 2.89%, average erythrocyte hemoglobin density 32.6 g/dL, and vitamin B12 839.65 pg/mL. Her blood glucose level was 212 mg/dL, the level of hemoglobin A1c was 7.3%, and the simultaneous measurement of serum level of C-peptide showed decreased level of 0.13 ng/mL. Serum concentration of total calcium were 5.5 mg/dL (range, 8.7 to 1.4); ionized Ca 1.56 mg/dL (range, 2.3 to 2.58); phosphorus 6.7 mg/dL (range, 2.5 to 4.5); total protein 6.7 g/dL; albumin 3.3 g/dL; alkaline phosphatase 329 IU/L; aspartate aminotransferase 119 IU; alanine aminotransferase 65 IU/L. Intact PTH level was 13.32 pg/mL (range, 13 to 54). Serum adrenocorticotropic hormone (ACTH) and cortisol levels were 11.92 pg/mL (range, 10 to 60) and 0.94 (range, 9.4 to 26.1). The results of rapid ACTH stimulation test for adrenal function were normal. The results of thyroid function test were T3 99.01 ng/mL, free T4 0.85 µg/dL, and thyroid stimulating hormone 7.99 µIU/mL. The results for antimicrosomal and antithyroglobulin antibody were positive (52.91 IU/mL, 132.57 IU/mL), and, both islet cell antibody and antiglutamic acid decarboxylase antibody tested negative. In the Ellsworth-Howard examination, urinary phosphoric acid was slightly increased (12.3 mg/dL) and the increase of cyclic adenosine monophosphate (cAMP) was sufficient (4,400 µmol/mL). There was no GNAS mutation. Serum levels of sodium, potassium, blood urea nitrogen, and creatinine were within normal limits at 144 mmol/L, 5.3 mmol/L, 8.2 mg/dL, and 1.0 mg/dL.
- Radiological results
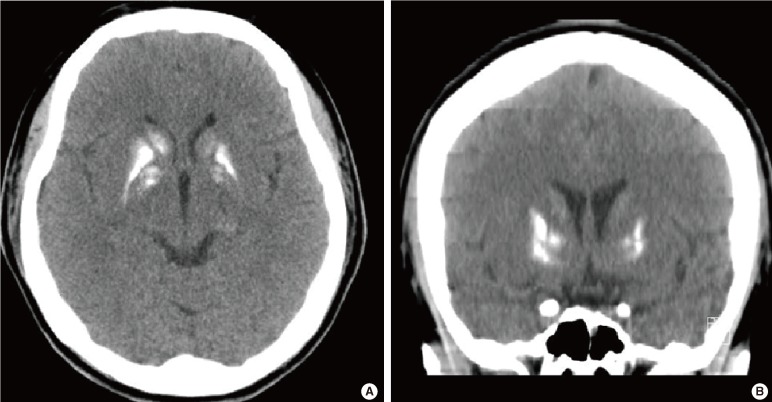
- The chest X-ray showed no abnormal lesion. An brain computed tomography scan showed prominent calcifications in both basal ganglion and hypothalamus (Fig. 1). The thyroid sonogram discovered several nodules, sized 4.5×3.7 mm and 3.9×2.9 mm on each side that were suspected to be thyroid papillary cancer.
- Treatment and clinical course
- The patient came to the hospital experiencing general weakness, and she was diagnosed with hypocalcaemia on laboratory examination. At that time, both the levels of PTH and vitamin D were within normal range, but she had previously experienced spasm symptoms and the levels of previous intact PTH were low (4.38 to 10.66 pg/mL) 7 years ago. She has taken oral calcium medicine for treatment of idiopathic hypoparathyroidism because she had no underlying secondary disorders for hypocalcemia and low PTH level. Although she has no pathognomonic brachydactyly, her appearance was similar to AHO in clinical features including short stature, intelligence decline, round face, calcification on basal ganglion and hypothalamus, or obesity. Furthermore, she had a lower normal PTH level and hypocalemia on admission, and we decided to investigate the PTH resistance to identify PHP Ia or PPHP with AHO. In the Ellsworth-Howard examination for investigation of the PTH resistance, it was confirmed that there was no PTH resistance at the kidney and there was no sign of GNAS mutation. We diagnosed this case as hypocalcemia due to idiopathic hypoparathyroidism for lower serum calcium and traced intact PTH levels and began calcium and calcitriol medicine to treat the hypocalcaemia. Subsequently, her symptoms were improved with normalization of ionized Ca at 2.32 mg/dL (range, 2.3 to 2.53). The results of fine needle aspiration on two nodules of thyroid were suggestive malignancy and she underwent a total thyroidectomy. The biopsy results were papillary microcarcinoma with lymphocytic thyroiditis for two suspicious nodules (sized 1.5×1.0 cm on the left and 2.0×1.0 cm on the right). Among the four parathyroid glands, the left inferior parathyroid gland was not discovered, and the remaining three parathyroid glands were remained during surgery. We used levothyroxine 0.05 mg medication for Hashimoto thyroiditis and insulin determir 12 U and aspart 16 U for glucose control.
CASE REPORT
- PAS was reported by Schmidt in 1922 when he conducted an autopsy of a patient who had died of adrenal insufficieny and discovered that the lymphocyte had infiltrated into the thyroid and adrenal cortex, and thereafter, many cases have been reported in which both autoimmune endocrinopathy and nonendocrinopathy have been found in the same patient. In 1980, Neufeld et al. [1] classified the PAS into type I, II, and III according to age of endocrinopathy occurrence, heredity method, relationship with HLA, and accompanying disease, and this contributed greatly to screening and early diagnosis of PAS. PAS type I is also called autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy, and it has been the topic of recent genetic research. This revealed that PAS type I is known to be associated with the abnormality of a single gene such as autoimmune regulator gene (AIRE) through autosomal recessive heredity [13,14]. In addition, the immune dysfunction, polyendocrineopathy, enteropathy, X-Linked (IPEX) syndrome is casued by serious immune damage with forkhead box P3 gene mutation at the regulatory T cell. PAS may be classified into three types including type I related with AIRE, type II related with HLA, and type III realted with IPEX. Particularly, PAS type I is so rare that only 100 cases have been reported worldwide [3].
- In Korea, some cases of PAS with various clinical features have been reported [4-8], but the type III case is not common. Seven cases of PAS type III have been reported; three cases of PAS with autoimmune thyroid disease and systemic lupus erythematosis, two cases with autoimmune thyroid disease and rheumatoid arthritis, one case with pernicious anemia and insulin-dependent diabetes, and one case with autoimmune thyroiditis and leukopathy. The patient in the present case was diagnosed with type I diabetes (C-peptide 0.13) accompanied by insulin deficiency 11 years ago and thyroiditis was already under development according to thyorid ultrasonogram examination that was taken 7 years ago. After then, antoimmune thyroiditis was confirmed with the positive results of the thyroid autoantibodies and biopsy. Therefore the patient was diagnosed with PAS III with autoimmune thyroid disease and type 1 diabetes.
- Primary hypoparathyroidism is characterized as a deficiency or secretory disorder of PTH without underlying disorders, including prior neck surgery, chronic renal failure, alcoholism, gastrointestinal absorption disorders, and pathognomonic musculoskeletal abnormality as stated by Albright [12]. AHO is known to be relatively rare in primary hypoparathyroidism, but one case with AHO and primary hypoparathyroidism has been reported in Korea [15]. AHO is a congenital disease associated with short stature, obesity, rounded face, subcutaneous ossification, and characteristic shortening and widening of long bones in the hands and feet, and less frequently, mental retardation [9,10]. Usually, AHO is associated with PHP I or PPHP with GNAS mutation [11]. Six cases of PPHP or PHP with AHO and two cases of PPHP or PHP without AHO have been reported in Korean [16,17]. The PPHP is considered when a patient has definite AHO evidences, a family history of PPHP, normal results on the Ellsworth-Howard examination, reductions in erythrocyte membrane Gs activity, GNAS mutation or a patient's father has AHO evidences [18,19]. The AHO patients with paternal inheritance may be diagnosed with PPHP without hormone resistance of PTH, but the patients with maternal inheritance may have PHP by hormone resistance [11]. The patient in the present case was confirmed with hypocalcemia, low PTH, and a previous history of spasms that might be asoociated with hypocalcemia. Basen on laboratory results, we could consider this case as primary hypoparathyroidism. Interistingly, she had several clinical features similar to AHO including short stature, intelligence decline, round face, calcification on basal ganglion and hypothalamus, and obesity except pathognomonic brachydactyly, which might be considered as PHP or PPHP. Therefore, the Ellsworth-Howard examination was performed to investigate the resistance to PTH actions on target organs [20] but we diagnosed the patient with idiopathic hypoparathyroidism due to the increase of urinary phosphoric acid, sufficient increase of cAMP by 20 times (4,400 µmol/mL), and no GNAS mutation. The subnormal response of urinary phosphoric acid might be due to variations of the basal level or diurnal execretion.
- In conclusion, it is thought that this is a case of PAS type IIIa with autoimmune thyroiditis and type 1 diabetes required insulin treatment. At the same time, this case was accompanied by thyroid papillary cancer and primary hypoparathyroidism with clinical features that could be mistaken for AHO. This is a rare case and instructive experience for us and we report the case with the review of reference literature.
- In the present case, the patient was young female who was diagnosed with type I diabetes as an absolute deficiency of insulin and required insulin supply for glucose control. She had also autoimmune thyroid disease, thyroid papillary cancer and primary hypoparathyroidism with clinical features resembled AHO. Here, we report a rare case of PAS type IIIa with autoimmune thyroiditis and type 1 diabetes, accompanied by thyroid papillary cancer and primary hypoparathyroidism with clinical features that could be mistaken for AHO.
DISCUSSION
- 1. Neufeld M, Maclaren N, Blizzard R. Autoimmune polyglandular syndromes. Pediatr Ann 1980;9:154–162. ArticlePubMed
- 2. Papadopoulos KI, Hallengren B. Polyglandular autoimmune syndrome type II in patients with idiopathic Addison's disease. Acta Endocrinol (Copenh) 1990;122:472–478. ArticlePubMed
- 3. Park YS. Loss of function of the autoimmune regulator (AIRE) gene and development of poyendocrinopathy. J Korean Soc Endocrinol 2003;18:439–449.
- 4. Song CU, Shong MH, Kim YK, Ro HK, Yoon SI, Kim SS, Seong KY, Kim SY. A case of polyglandular autoimmune syndrome presenting Graves' disease, pernicious anemia and insulin dependent diabetes mellitus. J Korean Soc Endocrinol 1993;8:211–216.
- 5. Jeong ET, Park JH, Kim DH, Jeong BO, Ko KS, Rhee BD. A case of polygrandular autoimmune syndrome. J Korean Soc Endocrinol 1995;10:418–423.
- 6. Kim CH, Kim HK, Park JY, Song YK, Kim KS, Yoo KS. A case of polyglandular autoimmune syndrome. J Korean Soc Endocrinol 1997;12:672–676.
- 7. Kim JS, Lee MD, Kim HS, Chung CH. A case of polygrandular automune type II syndrome associated empty sella. J Korean Soc Endocrinol 1998;13:295–300.
- 8. Lee YS, Lee JM, Park HO, Park SK, Yoon SR, Kim SY, Cha BY, Lee KW, Son HY, Kang SK. A case of type II autoimmune polyglandular syndrome: acute adrenal crisis presented as the first manifestation of Addison's disease in a patient with diabetic ketoacidosis and hypgonadism. J Korean Soc Endocrinol 1998;13:115–120.
- 9. Levine MA, Germain-Lee E, Jan de Beur S. Genetic basis for resistance to parathyroid hormone. Horm Res 2003;60(Suppl 3):87–95. ArticlePubMed
- 10. Lemay M, McNeely WF, Raisz LG. Dyschondroplasia with soft tissue calcification and ossification, and normal parathyroid function (pseudo-pseudohypoparathyroidism). Am J Med 1956;21:649–656. ArticlePubMed
- 11. Hayward BE, Moran V, Strain L, Bonthron DT. Bidirectional imprinting of a single gene: GNAS1 encodes maternally, paternally, and biallelically derived proteins. Proc Natl Acad Sci U S A 1998;95:15475–15480. ArticlePubMedPMC
- 12. Adams RD, Victor M. Principles of neurology; 4th ed. New York: McGraw-Hill Information Services Co.; 1989.
- 13. Nagamine K, Peterson P, Scott HS, Kudoh J, Minoshima S, Heino M, Krohn KJ, Lalioti MD, Mullis PE, Antonarakis SE, Kawasaki K, Asakawa S, Ito F, Shimizu N. Positional cloning of the APECED gene. Nat Genet 1997;17:393–398. ArticlePubMedPDF
- 14. Finnish-German APECED Consortium. An autoimmune disease, APECED, caused by mutations in a novel gene featuring two PHD-type zinc-finger domains. Nat Genet 1997;17:399–403. ArticlePubMed
- 15. Kim JK, Byun YJ, Park CS. A case of idiopathic hypoparathyroidism with Albright's hereditary osteodystrophy. J Korean Neurol Assoc 1991;9:349–356.
- 16. Ahn H, Kim KS, Kim IH, Song HJ, Cheon HW, Lee JW, Oh CH. A case of cutaneous ossification occurring in pseudohypoparathyroidism. Ann Dermatol 1999;11:263–266.Article
- 17. Kim SR, Doh YJ, Kim HK, Moon SS, Lee JY, Jeon JH, Kim SW, Kim BW, Lee IK, Kim JG. A case of pseudopseudohypoparathyroidism with normal stature. J Korean Endocr Soc 2009;24:138–143.Article
- 18. Weinstein LS, Yu S, Warner DR, Liu J. Endocrine manifestations of stimulatory G protein alpha-subunit mutations and the role of genomic imprinting. Endocr Rev 2001;22:675–705. PubMed
- 19. Levine MA, Jap TS, Mauseth RS, Downs RW, Spiegel AM. Activity of the stimulatory guanine nucleotide-binding protein is reduced in erythrocytes from patients with pseudohypoparathyroidism and pseudopseudohypoparathyroidism: biochemical, endocrine, and genetic analysis of Albright's hereditary osteodystrophy in six kindreds. J Clin Endocrinol Metab 1986;62:497–502. ArticlePubMedPDF
- 20. Ogata E, Yamamoto M, Matsumoto T, Fujita T, Fukase M, Kinoshita Y, Furukawa Y, Sohn HE, Nakajima H, Yasuda T. Standard procedure and the diagnostic criteria for the Ellsworth-Howard test using human PTH-(1-34). Nihon Naibunpi Gakkai Zasshi 1984;60:971–984. ArticlePubMed
References

Figure & Data
References
Citations
Citations to this article as recorded by 
- Autoimmune polyglandular syndrome type III associated with antineutrophil cytoplasmic autoantibody-mediated crescentic glomerulonephritis
Shiyuan Tian, Baofeng Xu, Ziwei Liu, Rui Liu
Medicine.2020; 99(7): e19179. CrossRef - Hypoparathyroidism: Genetics and Diagnosis
Michael Mannstadt, Luisella Cianferotti, Rachel I Gafni, Francesca Giusti, Elizabeth Helen Kemp, Christian A Koch, Kelly L Roszko, Liam Yao, Gordon H Guyatt, Rajesh V Thakker, Weibo Xia, Maria-Luisa Brandi
Journal of Bone and Mineral Research.2020; 37(12): 2615. CrossRef - A rare case of polyglandular autoimmune syndrome type IIIc with primary antibody failure
Adina Elena Stanciu, Florentina Sava, Gergely Toldi
Gynecological Endocrinology.2018; 34(4): 283. CrossRef - Autoimmune Polyendocrine Syndrome Type IIIC and Ankylosing Spondylitis; a Case Report
Ali Javinani, Hamid Reza Aghaei Meybodi, Ahmad Reza Jamshidi, Farhad Gharibdoost, Hoda Kavosi
Rheumatology Research.2016;[Epub] CrossRef - Brief Review of Articles in 'Endocrinology and Metabolism' in 2013
Won-Young Lee
Endocrinology and Metabolism.2014; 29(3): 251. CrossRef
 PubReader
PubReader Cite
Cite