Selective Mitochondrial Uptake of MKT-077 Can Suppress Medullary Thyroid Carcinoma Cell Survival In Vitro and In Vivo
Article information
Abstract
Background
Medullary thyroid carcinoma (MTC) is a neuroendocrine tumor mainly caused by mutations in the rearranged during transfection (RET) proto-oncogene. Not all patients with progressive MTC respond to current therapy inhibiting RET, demanding additional therapeutic strategies. We recently demonstrated that disrupting mitochondrial metabolism using a mitochondria-targeted agent or by depleting a mitochondrial chaperone effectively suppressed human MTC cells in culture and in mouse xenografts by inducing apoptosis and RET downregulation. These observations led us to hypothesize that mitochondria are potential therapeutic targets for MTC. This study further tests this hypothesis using1-ethyl-2-[[3-ethyl-5-(3-methylbenzothiazolin-2-yliden)]-4-oxothiazolidin-2-ylidenemethyl] pyridinium chloride (MKT-077), a water-soluble rhodocyanine dye analogue, which can selectively accumulate in mitochondria.
Methods
The effects of MKT-077 on cell proliferation, survival, expression of RET and tumor protein 53 (TP53), and mitochondrial activity were determined in the human MTC lines in culture and in mouse xenografts.
Results
MKT-077 induced cell cycle arrest in TT and MZ-CRC-1. Intriguingly, MKT-077 also induced RET downregulation and strong cell death responses in TT cells, but not in MZ-CRC-1 cells. This discrepancy was mainly due to the difference between the capacities of these cell lines to retain MKT-077 in mitochondria. The cytotoxicity of MKT-077 in TT cells was mainly attributed to oxidative stress while being independent of TP53. MKT-077 also effectively suppressed tumor growth of TT xenografts.
Conclusion
MKT-077 can suppress cell survival of certain MTC subtypes by accumulating in mitochondria and interfering with mitochondrial activity although it can also suppress cell proliferation via other mechanisms. These results consistently support the hypothesis that mitochondrial targeting has therapeutic potential for MTC.
INTRODUCTION
Medullary thyroid carcinoma (MTC) is a neoplasm of the endocrine system, which originates from parafollicular C-cells of the thyroid gland [1]. MTC is relatively rare, accounting for approximately 5% of all thyroid cancers, and progresses slowly. Nevertheless, MTC can be fatal and the only curative therapy is surgical resection, which is not effective for metastatic or recurring MTC. MTC occurs either sporadically or in hereditary forms, i.e., familial MTC and multiple endocrine neoplasia type 2 syndrome. Development of MTC is mainly attributed to various mutations in the receptor tyrosine kinase, rearranged during transfection (RET), although other oncogenic mutations (e.g., Ras mutations) are also detected in MTC at relatively low frequencies [234]. RET activating mutations occur mainly in the extracellular cysteine-rich receptor domain or the intracellular tyrosine kinase domain and these mutations are detected in about 95% of hereditary MTC and about 50% of sporadic MTC cases [5]. Therefore, RET is a key therapeutic target in MTC. Indeed, vandetanib (trade name Caprelsa, AstraZeneca, London, UK) and cabozantinib (Exelixis, South San Francisco, CA, USA), multi-tyrosine kinase inhibitors targeting RET and other tyrosine kinase receptors, have been recently approved for the therapy of inoperable progressive MTC [67]. Nevertheless, not all patients respond to these drugs, requiring the development of additional therapeutic strategies [678].
It is now well understood that mitochondrial metabolism is often reprogrammed in order to facilitate proliferation and survival of tumor cells. For example, mitochondrial oxidative phosphorylation in cancer is critical to meet increased demands for the production of building blocks required for uncontrolled tumor cell proliferation [9]. Moreover, altered levels of certain metabolic byproducts from mitochondria, including reactive oxygen species (ROS), have been implicated in tumor initiation, maintenance, as well as suppression [10]. Accordingly, targeting a mitochondrial metabolic alteration in cancer is emerging as a potential therapeutic strategy [1112], and it is important to determine whether this strategy can be an option for MTC therapy. Indeed, we have recently demonstrated that the mitochondria-targeted metabolic interfering agent, triphenyl-phosphonium-carboxy-proxyl (Mito-CP), or RNA interference of mortalin (GRP75/HSPA9), a mitochondrial chaperone of HSP70 family, can effectively suppress human MTC cells in culture and in mouse xenografts by inducing not only growth arrest but also substantial cell death [1314]. Of note, these treatments commonly induced cytotoxic effects in MTC cells by altering mitochondrial bioenergetics and ROS levels, which were accompanied by RET downregulation [1314]. These findings suggest that human MTC cells require mitochondrial activity for their survival and proliferation, leading to a hypothesis that mitochondria are effective targets for MTC therapy.
1-Ethyl-2-[[3-ethyl-5-(3-methylbenzothiazolin-2-yliden)]-4-oxothiazolidin-2-ylidenemethyl] pyridinium chloride (MKT-077) is a water-soluble lipophilic cationic rhodocyanine dye, which tends to accumulate in mitochondria via mitochondrial membrane potential (Δψm) [15]. Because its accumulation interferes with mitochondrial activity, leading to cell damages, and tumor cells generally maintain higher electrochemical gradient of Δψm, it has been proposed that MKT-077 accumulation and subsequent cell damages can occur preferentially in tumor cells [1516]. This property of MKT-077 was confirmed in a clinical setting although it did not pass the human clinical trials due to its high renal toxicity [17]. Of note, MKT-077 is also known for its ability to inhibit a few HSP70 family members including mortalin and HSC70 [18]. Given these properties, MKT-077 may be used to further determine whether mitochondria are effective therapeutic targets for MTC.
In this study, we have evaluated MKT-077 for its potential to suppress MTC cells and investigated the mechanism underlying its effects in MTC cells. In comparison with the U.S. Food and Drug Administration approved drug, vandetanib, we demonstrate that MKT-077 can suppress human MTC cell proliferation and survival in vitro as well as in mouse xenografts. We also demonstrate that the degree of MKT-077 toxicity in MTC cells is mainly determined by cellular levels of Δψm and is correlated with disrupted mitochondrial integrity. Our data consistently support the hypothesis that mitochondria have potential as a therapeutic target for MTC.
METHODS
Cell culture and reagents
The human MTC lines, TT and MZ-CRC-1, were maintained as previously described [192021]. Briefly, TT was maintained in RPMI 1640 (Invitrogen, Carlsbad, CA, USA) supplemented with 16% fetal bovine serum (FBS), 100 units of penicillin and 100 µg of streptomycin per mL. MZ-CRC-1 was maintained in high-glucose DMEM (Invitrogen) supplemented with 10% FBS in culture dishes coated with rat collagen (Sigma, St. Louis, MO, USA). All experiments were performed using cells within 10 passages from the point of acquisition. MKT-077, tetra-methyl-rhodamine ethyl ester perchlorate (TMRE), and N-acetyl-cysteine (NAC) were purchased from Sigma. 5-(and-6)-carboxy-2',7'-dichlorodihydrofluorescein diacetate (carboxy-H2DCFDA) and vandetanib were purchased from Invitrogen and LC Laboratories (Woburn, MA, USA), respectively.
MTT assay
The colorimetric 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyltetrazolium bromide (MTT; Sigma) assay was performed as previously described [22]. Briefly, cells were seeded in 24 well-plates and allowed to attach for 48 hours. After drug treatment, cells were incubated with 400 µL of MTT (0.5 mg/mL) in complete medium for 2 hours at 37℃, switched into 200 µL of dimethyl-sulfoxide (DMSO), and shaken for 5 minutes at room temperature before measuring absorbance at 540 nm.
Cell cycle analysis
Cells were washed in ice cold phosphate-buffered saline (PBS), fixed in 70% ethanol at -20℃, stained with Hoechst 33342 (1 µg/mL) in 3.8 mmol/L sodium citrate/PBS containing RNAase A (0.5 mg/mL) for 2 hours on ice, and analyzed by LSR-II flow cytometer (Becton Dickinson, San Jose, CA, USA) with a gate that selects single cells within a normal size range. Cell cycle parameters from 20,000 gated cells were determined and analyzed using FCS Express software (De Novo Software, Los Angeles, CA, USA).
Visualization of MKT-077 uptake
Cells were incubated with 1 µM MKT-077 and 100 nM Mitotracker Green FM (Thermo Fisher Scientific, Carlsbad, CA, USA) in culture medium for 30 minutes at 37℃ in the dark, washed with PBS, switched into phenol-red free medium before visualizing fluorescence under a microscope. Pictures were acquired and processed with MetaVue software (Molecular Devices, Sunnyvale, CA, USA). For flow cytometric measurement, MKT-077-treated cells were resuspended in 0.1% bovine serum albumin/PBS and analyzed by flow cytometry (PE channel, 575 nm). Data from 20,000 cells were analyzed using FCS Express software (De Novo Software).
Detection of mitochondria membrane potential and oxidative stress
To determine Δψm, 2×105 cells were plated per well in 6-well plates and incubated for 24 hours. Cells were then incubated with 5 nM TMRE for 15 minutes and analyzed by flow cytometry, as previously described [13]. To detect ROS generation, cells were incubated with the drugs for 24 hours, and then cells were treated with 1 µM carboxy-H2DCFDA in a drug-free culture medium at 37℃ for 1 hour. After medium change, cells were incubated for 2 hours in the dark, trypsinized, and resuspended in PBS, before analysis by flow cytometry (fluorescein isothiocyanate channel, 525 nm). Data from 20,000 cells were analyzed using FCS Express software (De Novo Software).
Viral infection and RNA interference
Tumor protein 53 (TP53) was depleted using pLKO.1 lentiviral shRNA systems, which were previously described [23]. For lentivirus production, 293T cells were co-transfected with pLKO.1 and packaging vectors, as previously described [24]. Viral supernatants were collected after 72 hours and mixed with polybrene (Sigma) at 4 to 8 µg/mL before use. Viral titer was determined by scoring cells expressing green fluorescence protein. Specific knockdown of target proteins was confirmed by Western blot analysis.
Immunoblot analysis
Cells were lysed in 62.5 mmol/L Tris (pH 6.8)-2% sodium dodecyl sulphate (SDS) mixed with the protease inhibitor cocktail (Sigma) and briefly sonicated before determining the protein concentration using the BCA reagent (Pierce, Rockford, IL, USA). A 50 µg of protein was resolved by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a polyvinylidene difluoride membrane filter (Bio-Rad, Hercules, CA, USA). Membrane filters were blocked in 0.1 M Tris (pH 7.5)-0.9% NaCl-0.05% Tween 20 with 5% nonfat dry milk, and incubated with appropriate antibodies. Antibodies were diluted as follows: glyceraldehyde-3-phosphate dehydrogenase (GAPDH), 1:5,000; poly(ADP-ribose) polymerase (PARP), 1:1,000; cleaved lamin A, 1:2,000; β-actin, 1:10,000; cytochrome c oxidase (COX IV), 1:2,000 (Cell Signaling, Danvers, MA, USA); RET, 1:1,000; TP53, 1:1,000; GRP75, 1:2,000 (Santa Cruz Biotechnology, Santa Cruz, CA, USA); E2 promoter binding factor 1 (E2F-1), 1:1,000 (NeoMarkers, Fremont, CA, USA); Ki-67, 1:1,000 (Dako, Glostrup, Denmark); peroxisome proliferator-activated receptor gamma coactivator 1α (PGC-1α), 1:2,000 (Abcam, Cambridge, UK). The Supersignal West Femto and Pico chemiluminescence kits (Pierce) were used for visualization of the signal. Images of immunoblots were taken and processed using ChemiDoc XRS+ and Image Lab 3.0 (Bio-Rad).
Tumor xenograft studies
The 1×107 TT cells in 200 µL Hank's balanced salt solution were inoculated subcutaneously into the rear flanks of 6-week-old female athymic nude (nu/nu) mice (Charles River Laboratories, Wilmington, MA, USA). Once palpable, tumors were measured using Vernier calipers at intervals indicated in the text. Tumor volumes (TVs) were calculated using the formula: TV=L×W2×0.5 (L, length; W, width). When TV reached 100 mm3, mice were sorted into groups of 8 to achieve equal distribution of tumor size in all treatment groups. Group 1 received only the vehicle (1:9 mixture of DMSO/saline) and group 2 received MKT-077 (10 mg/kg body weight/dose). A 200 µL of ether solution was administered by intraperitoneal injection every 2 days (total 10 doses). At the end of the experiments, animals were euthanized by CO2 asphyxiation. All animal studies were performed according to protocols approved by the Institutional Animal Care and Use Committee at Medical College of Wisconsin.
Statistical analysis
Two-tailed unpaired Student t test was used to assess the statistical significance of two data sets. P value <0.05 was considered statistically significant.
RESULTS
MKT-077 induces differential growth inhibitory effects in human MTC cells
To determine whether MKT-077 can induce growth inhibitory effects in MTC cells, we used the two most thoroughly characterized human MTC lines, TT and MZ-CRC-1. TT cells harbor RETC634W mutation in the extracellular domain of RET whereas MZ-CRC-1 cells harbor RETM918T in the RET kinase domain. These cell lines have been extensively used for the evaluation of different agents for MTC therapy [252627]. As determined by MTT assay, MKT-077 treatment (0.1 to 10 µM dose ranges) for 48 hours could effectively decrease TT cell viability, which was quite comparable to the effects of vandetanib used at equivalent dosage (Fig. 1A). In contrast, MKT-077 treatment was not as effective as vandetanib in MZ-CRC-1 cells although the agent suppressed MZ-CRC-1 cell viability when used at higher doses (Fig. 1A). The calculated IC50 values under these conditions were 0.74 µM for TT and 11.4 µM for MZ-CRC-1 respectively. Therefore, TT cells were more sensitive to MKT-077 than MZ-CRC-1 cells.
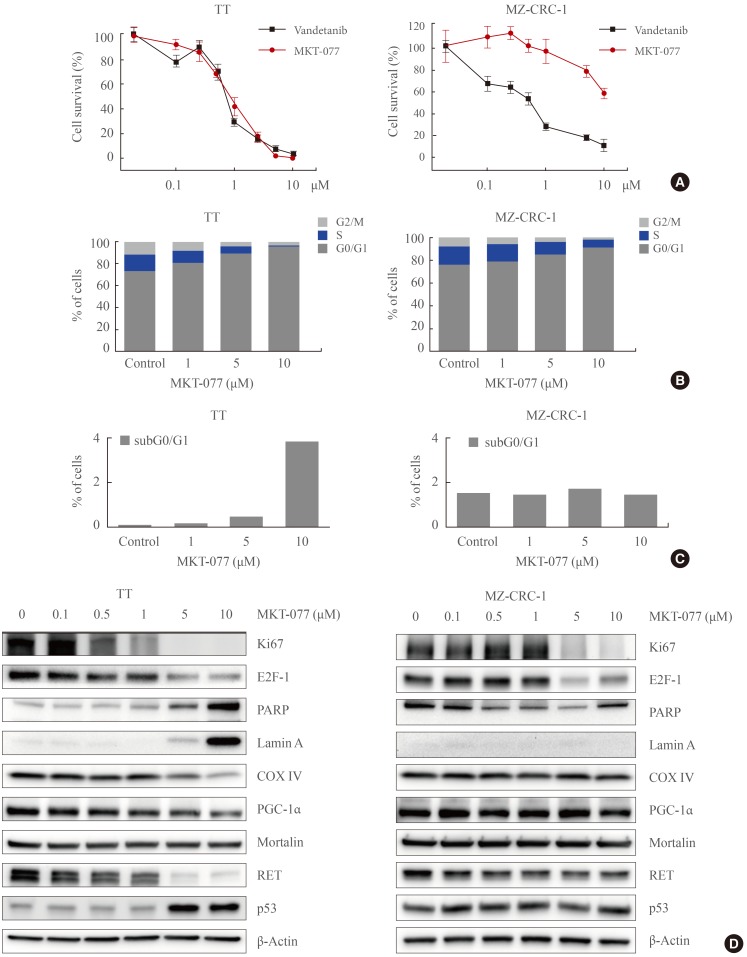
1-Ethyl-2-[[3-ethyl-5-(3-methylbenzothiazolin-2-yliden)]-4-oxothiazolidin-2-ylidenemethyl] pyridinium chloride (MKT-077) induces growth inhibitory effects in medullary thyroid carcinoma (MTC) cell culture. (A) TT and MZ-CRC-1 cells in 24-well plates were treated with serially increasing doses of MKT-077 and vandetanib for 48 hours. Cells were then allowed to recover in drug-free fresh medium for 48 hours, prior to measurement of cell viability by MTT assay. Data (mean±SD, n=6) are expressed as the percentage of vehicle-treated control. (B, C) TT and MZ-CRC-1 cells were treated with MKT-077 at indicated doses for 48 hours prior to cell cycle analysis using Hoechst 33342. Control cells were treated with equal volume of dimethyl sulfoxide. (B) Data are expressed as the percentage of living cells in each cell cycle phase. (C) Number of apoptotic cells was estimated by counting cells in the subG0/G1 phase. (D) Total lysates of TT and MZ-CRC-1 cells treated with different doses of MKT-077 for 48 hours were analyzed by Western blotting for indicated proteins. β-Actin is the control for equal loading. Ki67, MKI67-encoded protein; E2F-1, E2 promoter binding factor 1; PARP, poly(ADP-ribose) polymerase; COX IV, cytochrome c oxidase; PGC-1α, peroxisome proliferator-activated receptor gamma coactivator 1α; RET, rearranged during transfection.
To understand the nature of MKT-077-induced growth suppressive effects in TT and MZ-CRC-1 cells, we conducted cell cycle analyses using Hoechst 33342. MKT-077 treatment resulted in accumulation of cells in the G0/G1 phase in a dose-dependent manner, indicating that MKT-077 induced cell cycle arrest in these cells (Fig. 1B). Consistent with the higher sensitivity of TT cells to MKT-077, cell cycle arrest was more prominent in TT cells (Fig. 1B). Of note, MKT-077 also increased sub-G0/G1 phase population in TT cell culture in a dose-dependent manner, suggesting that MKT-077 induced not only cell cycle arrest but also cell death in TT cell culture (Fig. 1C). However, an equal MKT-077 treatment did not increase sub-G0/G1 phase population in MZ-CRC-1 cell culture (Fig. 1C).
In agreement with its ability to induce cell cycle arrest, MKT-077 downregulated cellular levels of the proliferation maker, Ki67, and the S-phase transcription factor, E2F-1, in TT and MZ-CRC-1 cells (Fig. 1D). However, MKT-077 induced cleavage of PARP and lamin A, the markers of caspase-dependent apoptotic cell death [28], only in TT cells (Fig. 1D), which is consistent with the selective increases in sub-G0/G1 phase population by MKT-077 in TT cell culture. Of note, MKT-077 significantly decreased the levels of COX IV, a marker of mitochondrial integrity, in TT, but not in MZ-CRC-1, cells, suggesting that MKT-077 may induce mitochondrial stress in TT cells (Fig. 1D). Consistent with this possibility, MKT-077 also decreased, albeit mildly, cellular levels of PGC-1α, a key regulator of mitochondria biogenesis and function, in TT but not MZ-CRC-1 cells (Fig. 1D). However, MKT-077 did not affect the levels of the key mitochondrial chaperone mortalin (Fig. 1D), suggesting that the decreases in COX IV levels may reflect a specific stress rather than overall decreases in the mitochondrial contents. Along with these effects, MKT-077 induced RET downregulation and TP53 upregulation only in TT cells. These data demonstrate that MKT-077 can induce growth inhibitory effects in MTC cells by activating common as well as distinct mechanisms.
Differential mitochondrial accumulation of MKT-077 in MTC cells
MKT-077 is selectively accumulated in mitochondria because of its lipophilic cationic property sensitive to Δψm [1529]. The degree of its mitochondrial accumulation therefore can vary among different cell types [1630]. To understand why MKT-077 induced differential growth inhibitory effects on TT and MZ-CRC-1 cells, we determined whether MKT-077 was accumulated in these cells at a similar degree. Fluorescence microscopy in combination with Mitotracker Green FM, a Δψm-independent mitochondrial dye, visualized high intensity fluorescence of MKT-077 that overlapped with Mitotracker Green FM in TT cells (Fig. 2A), indicating efficient mitochondrial accumulation of MKT-077 in the cell line. In contrast, no significant MKT-077 fluorescence was detected in MZ-CRC-1 cells, although mitochondria were efficiently stained with the Mitotracker in the cells (Fig. 2A). Moreover, flow cytometry using different doses of MKT-077 revealed that TT cells can uptake and retain MKT-077 at significantly higher levels than MZ-CRC-1 cells (Fig. 2B, C). These different rates of MKT-077 accumulation detected in TT and MZ-CRC-1 cells are consistent with the different levels of MKT-077 toxicity observed in these cells. To address this discrepancy in MKT-077 uptake between TT and MZ-CRC-1 cells, we compared their basal Δψm using TMRE, a Δψm-dependent mitochondrial dye [31]. Indeed, significantly higher TMRE staining was observed in TT cells (Fig. 2D), although cellular levels of mortalin, COX IV and PGC-1α, the markers for mitochondrial integrity, did not show any significant difference between these two cell lines (Fig. 2E). Both cell lines also expressed abundant RET, while higher RET levels being detected in MZ-CRC-1 (Fig. 2E). These data suggest that TT cells maintain much higher basal Δψm than MZ-CRC-1 cells while possibly maintaining similar levels of mitochondrial integrity. Therefore, the higher accumulation of MKT-077 in TT cells is mainly attributed to the higher basal Δψm in TT cells.
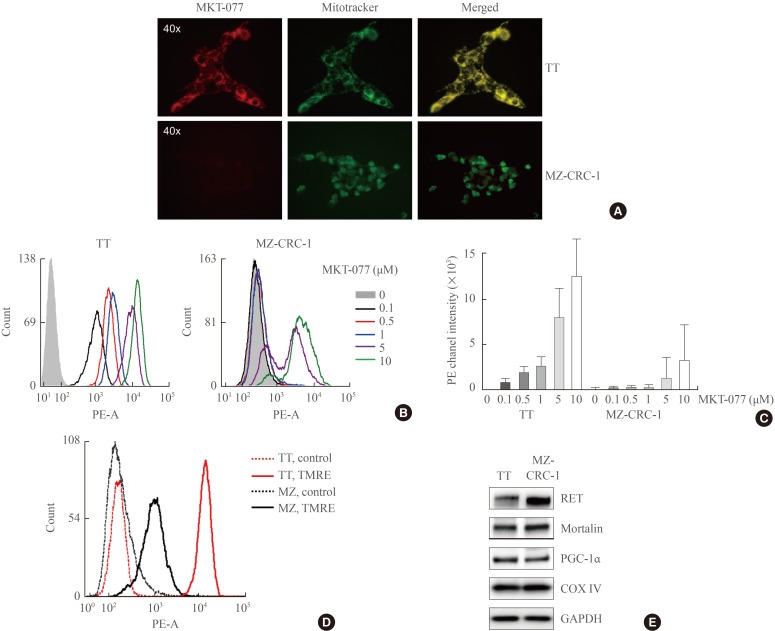
Differential mitochondrial accumulation of 1-ethyl-2-[[3-ethyl-5-(3-methylbenzothiazolin-2-yliden)]-4-oxothiazolidin-2-ylidenemethyl] pyridinium chloride (MKT-077) in medullary thyroid carcinoma (MTC) cells. (A) TT and MZ-CRC-1 cells were treated with 1 µM MKT-077 and 100 nM MitoTracker Green FM (Thermo Fisher Scientific) for 30 minutes. Pictures were then taken under a fluorescent microscope (×400). (B, C) TT and MZ-CRC-1 cells, treated with increasing doses of MKT-077 for 1 hour, were analyzed by flow cytometry to measure red fluorescence (PE channel, 575 nm) (B). (C) Data of 20,000 cells per dose were normalized to dimethyl sulfoxide-treated control and presented as mean±SD. (D) TT and MZ-CRC-1 cells were stained with 5 nM TMRE for 15 minutes and analyzed by flow cytometry to measure red fluorescence (PE channel, 575 nm). Unstained cells were used as negative controls. (E) Total lysates of TT and MZ-CRC-1 cells were analyzed by Western blotting for indicated proteins. GAPDH is the control for equal loading. TMRE, tetra-methyl-rhodamine ethyl ester perchlorate; RET, rearranged during transfection; PGC-1α, peroxisome proliferator-activated receptor gamma coactivator 1α; COX IV, cytochrome c oxidase; GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Cytotoxic effects of MKT-077 on TT cells are independent of TP53
It was previously shown that MKT-077 can activate TP53-mediated transcription by liberating the tumor suppressor from mortalin-mediated cytosolic sequestration [3233]. Given the pivotal requirement of mortalin for MTC cell survival [14], we first determined the effect of MKT-077 on the levels of mortalin in MTC cells and found that MKT-077 did not affect mortalin levels in TT and MZ-CRC-1 cells (Fig. 1D). Because TT cells harbor wild type TP53 and MKT-077 upregulated TP53 levels in these cells (Fig. 1D), we next determined the significance of TP53 for MKT-077-induced growth inhibitory effects using RNA interference. In TT cells, shRNA-mediated TP53 knockdown substantially blocked basal and MKT-077-induced TP53 levels (Fig. 3A). Under this TP53-depleted condition, we found that MKT-077 could still induce substantial downregulation of Ki67, E2F-1, and RET while inducing PARP cleavage without any significant difference compared to its effects in control cells (Fig. 3A). Consistent with these data, TP53 depletion could not also significantly inhibit MKT-077-induced viability loss in TT cell culture, as determined by MTT assay (Fig. 3B). These data suggest that TP53 is not required for MKT-077 to induce growth arrest and cell death in TT cells, but other mechanisms mediate MKT-077 toxicity.
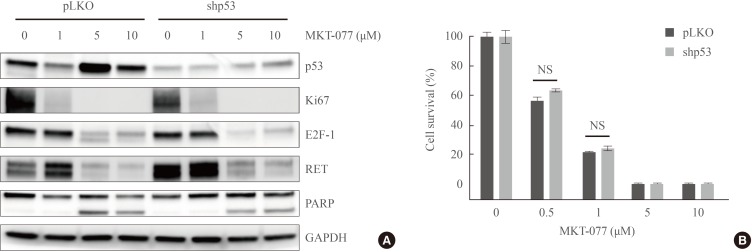
1-Ethyl-2-[[3-ethyl-5-(3-methylbenzothiazolin-2-yliden)]-4-oxothiazolidin-2-ylidenemethyl] pyridinium chloride (MKT-077) effects are mediated independently of TP53. (A) Western blot analysis of total lysates. TT cells were infected for 5 days with lentiviral pLKO.1 expressing TP53-targeting shRNA (shp53) or the control virus pLKO.1 prior to the treatment with indicated doses of MKT-077 for 48 hours. (B) TT cells infected with lentiviral shp53 or the control pLKO.1 in 24-well plates were treated with increasing doses of MKT-077 for 48 hours. Cells were then allowed to recover in drug-free fresh medium for 48 hours prior to the measurement of cell viability by MTT assay. Data (mean±SD, n=4) are expressed as the percentage of vehicle-treated control. Ki67, MKI67-encoded protein; E2F-1, E2 promoter binding factor 1; RET, rearranged during transfection; PARP, poly(ADP-ribose) polymerase; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; NS, not significant.
MKT-077 induces oxidative stress in TT cells
Mitochondrial damages activate cell death mechanisms [34], wherein oxidative stress caused by electron leakage from the respiratory chain is often involved [35]. To determine whether MKT-077 treatment increased the generation of ROS in TT cells, we measured cellular oxidation levels of carboxy-H2DCFDA, a redox-sensitive dye that fluoresces upon oxidation [36]. Within 24 hours of MKT-077 treatment, significantly increased fluorescence of carboxy-H2DCFDA was detected in TT cells in commensuration to MKT-077 doses (Fig. 4A).
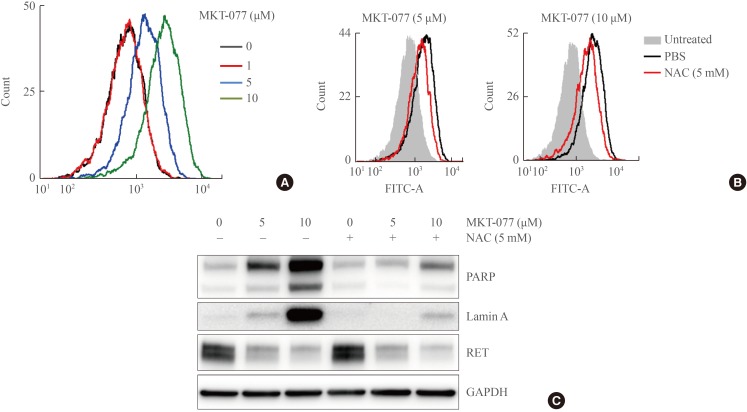
The reactive oxygen species (ROS) scavenger, N-acetyl-cysteine (NAC), can attenuate 1-ethyl-2-[[3-ethyl-5-(3-methylbenzothiazolin-2-yliden)]-4-oxothiazolidin-2-ylidenemethyl] pyridinium chloride (MKT-077)-induced ROS production and cell death, but not RET down-regulation. (A) TT cells treated with MKT-077 for 24 hours were treated with 1 µM 5-(and-6)-carboxy-2',7'-dichlorodihydrofluorescein diacetate (carboxy-H2DCFDA) for 1 hour and then incubated for 2 hours in a dye-free culture medium. Cells were harvested and analyzed by flow cytometry to measure green fluorescence (fluorescein isothiocyanate [FITC] channel, 525 nm). (B) Cells were pretreated with 5 mM NAC for 2 hours, and then incubated with indicated doses of MKT-077 for 24 hours before measurement of carboxy-H2DCFDA fluorescence by flow cytometry. (C) Cells were treated with indicated doses of MKT-077 in the presence of 5 mM NAC in the medium containing 2% fetal bovine serum (FBS) for 24 hours. Total cell lysates were analyzed by Western blotting for indicated proteins. GAPDH is the control for equal loading. PARP, poly(ADP-ribose) polymerase; RET, rearranged during transfection; GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Next, we determined whether the cell-permeable ROS scavenger, NAC could inhibit MKT-077-induced growth inhibitory effects on TT cells. First, we found that pretreatment with 5 mmol/L NAC significantly, albeit partially, reduced the levels of carboxy-H2DCFDA fluorescence in TT cells treated with different doses of MKT-077 (Fig. 4B). Consistent with these data, NAC pretreatment substantially reduced cleavage of PARP and lamin A, as determined by Western blotting (Fig. 4C). However, intriguingly, NAC pretreatment did not block MKT-077-induced RET downregulation (Fig. 4C). These data suggest that MKT-077 induces oxidative stress to suppress proliferation and survival of TT cells although it mediates RET downregulation independently of oxidative stress.
MKT-077 effectively suppresses TT xenografts in mice
We also determined whether MKT-077 could suppress TT xenografts in immune-compromised nude mice. Systemic administration of MKT-077 significantly delayed the growth of TT xenografts in mice throughout the treatment (Fig. 5A). At the end of the drug treatment, we found that tumor weights were about two-times less in MKT-077-treated group than in control group (Fig. 5B). These data are consistent with the growth inhibitory effects of MKT-077 observed in the in vitro setting above. Nonetheless, MKT-077 treatment resulted in weight loss and general toxicity in animals, which is consistent with its previously reported side effects in human patients [17].
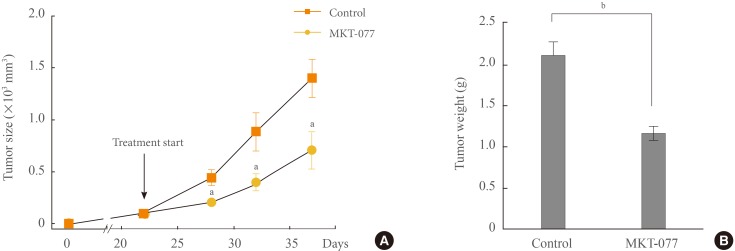
1-Ethyl-2-[[3-ethyl-5-(3-methylbenzothiazolin-2-yliden)]-4-oxothiazolidin-2-ylidenemethyl] pyridinium chloride (MKT-077) suppresses TT xenografts in athymic mice. (A) Athymic mice bearing TT xenografts were treated with MKT-077 (10 mg/kg/dose). Drug was administered intraperitoneal every two days beginning from day 22 after tumor implantation. The control group was treated with the vehicle only (METHODS). Changes in tumor sizes were determined at indicated time-points. (B) Weights of tumors collected at the end of the treatment. Data are mean±SE (n=8). aP<0.05; bP<0.001.
DISCUSSION
Although targeting oncogenic receptor tyrosine kinases using small molecule inhibitors has improved patient survival from surgically incurable metastatic or recurring MTC [68], substantial portion of progressive MTC cases are not responsive to the therapy, requiring additional therapeutic strategies. We previously reported that human MTC cells require mitochondrial activity for their survival and proliferation, which was determined by using mitochondrial targeting small molecule compound, Mito-CP, or by depleting mitochondrial chaperone, mortalin [1314]. These findings led us to hypothesize that mitochondria are an effective therapeutic target for MTC treatment. The present study consistently supports this hypothesis by demonstrating a strong correlation between mitochondrial accumulation of MKT-077 and its toxicity in MTC cells.
MKT-077 treatment could induce growth inhibitory effects more robustly in TT cells. Of note, MKT-077 induced RET downregulation in TT cells, which is very similar effects to the effects of Mito-CP or mortalin depletion in the cell line [13]. Together, these results strongly suggest that mitochondria have a role in regulating RET expression levels in MTC cells. Currently, relevant information is very limited to speculate by what mechanism(s) mitochondria can regulate RET expression in cells. Intriguingly, a very recent study has reported that RET has a protective role in maintaining the mitochondrial integrity in a drosophila model for neurodegenerative disease [37], supporting the existence of a link between RET and mitochondria. Because our previous [13] and current data exclude the potential involvement of oxidative stress in this regulation, a ROS-independent mechanism may be activated upon mitochondrial damage to mediate RET downregulation.
Our present study demonstrates that TP53 is not required for MKT-077 to mediate growth inhibitory effects. It has been reported that MKT-077 can interact with a few HSP70 family members, including mortalin and HSC70, and that these interactions are particularly important for liberating TP53 from the sequestration by these molecular chaperones and for subsequently activating the tumor suppressor [183238]. Although this accounts for a mechanism for tumor suppressive effects of MKT-077 in different tumor types, our data suggest that MKT-077 can also mediate tumor suppression via a mechanism(s) independent of TP53. It is possible that MKT-077 induces cytotoxic effects in TT cells by targeting TP53-independent mortalin function.
Intriguingly, MKT-077 did not induce cytotoxic effects in MZ-CRC-1 cells as effectively as in TT cells. This discrepancy appears to be mainly due to the inability of MZ-CRC-1 cells to uptake or retain MKT-077 in mitochondria as efficiently as TT cells do. Therefore, different MTC lines may have different responsiveness to MKT-077. This observation is somewhat similar to our previous observation [13], in which Mito-CP could induce cytotoxicity in TT cells more effectively than in MZ-CRC-1 cells in vitro as well as in vivo settings. Because mitochondrial delivery of these agents depends upon Δψm [1529], and their selective effects in tumors are expected based upon the notion that tumor cells generally maintain higher electrochemical gradient of Δψm [1529], these differential outcomes in TT and MZ-CRC-1 cells may suggest that evaluation of mitochondrial activity may predict MTC cell responsiveness to a therapeutic strategy that target mitochondria. In conclusion, our results consistently support the hypothesis that the mitochondria have potential as a therapeutic target for the treatment of MTC.
ACKNOWLEDGMENTS
This work was supported by American Cancer Society (RSGM-10-189-01-TBE) and the National Cancer Institute (R01CA138441) to JIP.
Notes
CONFLICTS OF INTEREST: No potential conflict of interest relevant to this article was reported.