Articles
- Page Path
- HOME > Endocrinol Metab > Volume 32(1); 2017 > Article
-
Review ArticleThe Role of Macrophage Lipophagy in Reverse Cholesterol Transport
-
Se-Jin Jeong
 , Mi-Ni Lee, Goo Taeg Oh
, Mi-Ni Lee, Goo Taeg Oh -
Endocrinology and Metabolism 2017;32(1):41-46.
DOI: https://doi.org/10.3803/EnM.2017.32.1.41
Published online: March 20, 2017
Immune and Vascular Cell Network Research Center, National Creative Initiatives, Department of Life Sciences, Ewha Womans University, Seoul, Korea.
- Corresponding author: Goo Taeg Oh. Immune and Vascular Cell Network Research Center, National Creative Initiatives, Department of Life Sciences, Ewha Womans University, 52 Ewhayeodae-gil, Seodaemun-gu, Seoul 03760, Korea. Tel: +82-2-3277-4128, Fax: +82-2-3277-3760, gootaeg@ewha.ac.kr
• Received: February 10, 2017 • Revised: February 20, 2017 • Accepted: February 27, 2017
Copyright © 2017 Korean Endocrine Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
- Macrophage cholesterol efflux is a central step in reverse cholesterol transport, which helps to maintain cholesterol homeostasis and to reduce atherosclerosis. Lipophagy has recently been identified as a new step in cholesterol ester hydrolysis that regulates cholesterol efflux, since it mobilizes cholesterol from lipid droplets of macrophages via autophagy and lysosomes. In this review, we briefly discuss recent advances regarding the mechanisms of the cholesterol efflux pathway in macrophage foam cells, and present lipophagy as a therapeutic target in the treatment of atherosclerosis.
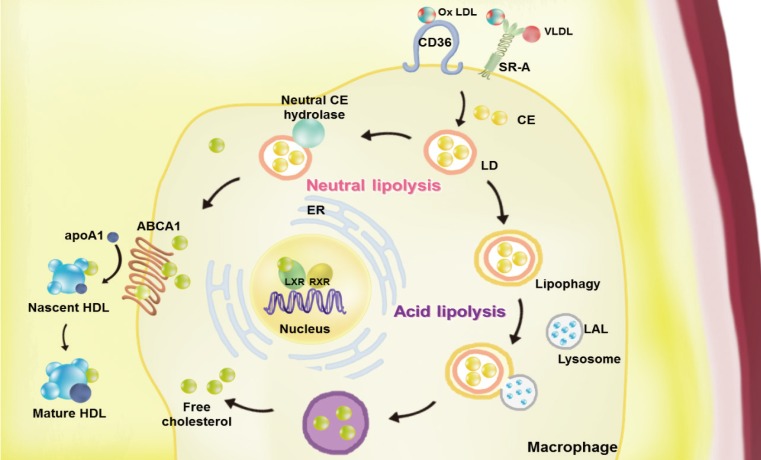
- Atherosclerosis is a chronic inflammatory disease characterized by the development of lipid-rich plaques that inhibit arterial blood flow [12]. Animal experiments and human specimen investigations have established that hypercholesterolemia promotes the inflammatory processes leading to atherosclerosis. Hypercholesterolemia induces the accumulation of apolipoprotein B (apoB)-rich lipoprotein, the main protein in atherogenic lipoprotein particles such as low density lipoprotein (LDL), very low density lipoprotein (VLDL), and lipoprotein(a), in the intima under the endothelial cell layer, leading to the recruitment of monocytes and initiation of the immune response. These monocyte-derived macrophages play an important role throughout the entire process of atherogenesis. Interestingly, recent studies have revealed that macrophage lipophagy has a novel function in contributing to the development of vascular disease. In this review, we discuss the role of macrophages in cholesterol metabolism in reverse cholesterol transport (RCT) and the contribution of macrophage lipophagy to atherosclerosis (Fig. 1).
INTRODUCTION
- RCT is a process through which excess cholesterol from peripheral cells and tissues returns to the liver for excretion, and plays an important role in reducing atherosclerosis. Macrophage cholesterol efflux is the first step of RCT that occurs in atherosclerotic vessel wall by macrophage-specific. In the early stages of atherogenesis, apoB-lipoproteins that have entered the intima are modified by processes such as oxidation and hydrolysis. These modifications lead to lipoprotein aggregation and further promote lipoprotein retention in the vessel wall [3]. The inflammatory signals originating from modified lipoproteins trigger endothelial activation and monocyte recruitment into the intima, and cause monocytes to differentiate into macrophages. Macrophages uptake modified lipoproteins within the cytoplasm through scavenger receptors (SRs), resulting in foam cell formation. Foam cell formation is the initial and key event of atherosclerosis.
- Macrophage scavenger receptors
- Although macrophages can clear modified LDL within intima through the low density lipoprotein receptor (LDLR), because the expression of LDLR is reduced in the early stage of foam cell formation by a decrease in sterol regulatory element-binding protein (SREBP)-1c due to increased cellular cholesterol levels [4], macrophages use another type of membrane receptor for apoB-lipoprotein removal. SRs are a diverse range of transmembrane proteins that internalize modified LDL and lipoprotein-base ligands. SRs have been divided into eight subclasses that share the defining feature of being able to bind various forms of modified LDL [5]; additionally, they can perform a wide variety of other functions, such as phagocytosis, antigen presentation, and elimination of apoptotic cells, depending on what they bind to. Nevertheless, the first and most important receptors that are responsible for modified LDL uptake in intimal macrophages are scavenger receptors type 1 (SR-A1) and cluster of differentiation 36 (CD36) [6].
- SR-A and CD36 are responsible for 75% to 90% of modified LDL degradation, and macrophages harvested from SR-A/CD36 double-null mice show an abnormal accumulation of cholesteryl esters (CEs) derived from modified LDL [7]. It has been evidently revealed in vitro that SR-A1 and CD36 play a crucial role in foam cell formation and LDL uptake in macrophages. However, the results of in vivo studies using gene-knockout models are somewhat different. The effect of SR-A1 on atherogenesis is controversial, whereas the proatherogenic role of CD36 has been clearly demonstrated in vivo. In 2005, Moore et al. [8] reported that defects in macrophage lipid uptake were found in apolipoprotein E (apoE) deficient mice in which either the CD36 or SR-A gene was deleted, leading to an increase in the size of atherosclerotic lesions. However, the other expanded studies showed that less atherosclerotic lesion formation in CD36 deficiency in apoE or LDLR null atherogenic mice model. Moreover, CD36 deficiency is sufficient to decrease of atherosclerosis in CD36/SR-A/apoE triple-null mice without the additional effect of SR-A1 deficiency [910]. Furthermore, a bone marrow transplantation assay also provided support for the proatherogenic role of macrophage CD36 [11]. These studies have demonstrated that macrophage CD36 promotes atherosclerosis via the uptake of modified LDL.
- Cholesterol esterification
- Within the macrophage, modified LDL is hydrolyzed to free cholesterol and fatty acid. Excess free cholesterol undergoes reesterification by the endoplasmic reticulum (ER)-resident protein acetyl-coenzyme A cholesterol acyltransferase 1 (ACAT1) or by sterol O-acyltransferase 1 (SOAT1), and is stored as CE in cytoplasmic lipid droplets (LDs). Although decreasing the expression or activity of ACAT1 was expected to have therapeutic effects through inhibition of foam cell formation, both ACAT1/apoE and ACAT1/LDLR double-null mice have been found to show similarly sized or only slightly smaller atherosclerotic lesions than controls [1213]. Furthermore, the efficacy of ACAT inhibitors in clinically preventing atherosclerosis in humans has not been successfully demonstrated [14]. Thus, these studies have clarified that cholesterol esterification is a passive protective response to excess free cholesterol when cholesterol efflux pathways are saturated.
MACROPHAGES IN REVERSE CHOLESTEROL TRANSPORT
- Neutral cholesterol lipolysis
- The hydrolysis of intracellular CE is the initial step of cholesterol efflux in macrophages. Because CE hydrolysis, which precedes cholesterol efflux, occurs in the cytoplasm at neutral pH levels, the catalyze enzymes have been collectively called neutral CE hydrolase. Three enzymes have been proposed to be components of neutral CE hydrolase in macrophages: hormone-sensitive lipase (HSL) [15]; carboxylesterase 1 (CEH or CES1), the human homolog of the murine triacylglycerol hydrolase [16]; and neutral cholesterol ester hydrolase 1 (NCEH1), which is also known as KIAA1363 or arylacetamide deacetylase like 1 (AADACL1) [17]. However, their importance has yet to be fully elucidated. Nevertheless, a common feature of all of the studies that have been conducted on this topic is that enhancing LD-related CE hydrolysis reduces CE accumulation and improves cholesterol efflux; thereby, reducing arteriosclerosis [1819]. Thus, these studies demonstrate that CE hydrolysis in macrophage cholesterol efflux is rate-limiting, and show that it is important to clarify the mechanisms mediating CE hydrolysis in foam cells for the treatment of atherogenesis.
- Acidic cholesterol lipolysis and lipophagy
- In 1999, Avart et al. [20] firstly discovered in vitro that a lysosomal component is involved in CE hydrolysis in foam cells. In this process, CE is hydrolyzed by lysosomal acid lipase (LAL), a lysosomal cholesterol esterase that exhibits optimal activity at the acidic pH of the lysosomal lumen. Recently, Ouimet et al. [21] reported that under lipid-loading conditions, autophagy mediated the delivery of cytoplasmic LDs to lysosomes in macrophages and that LAL in the lysosomal lumen hydrolyzed LD CE to generate free cholesterol for efflux. Ouimet et al. [21] also found autophagy to be specifically induced in response to atherogenic lipoprotein accumulation within macrophages, and thus, concluded that the autophagy-LAL pathway is a critical contributor to the mobilization of LD-associated CE for RCT.
- Autophagy is a conserved cellular process for the natural breakdown of unnecessary or non-functional cellular organelles and proteins by fusion with lysosomal compartments [22]. Autophagy is highly inducible under environmental stresses such as starvation and oxidative stress, and it plays an important role in maintaining essential cellular functions and protecting against infections with pathogens [2324]. In starvation conditions, autophagy promotes the degradation of cytoplasmic components non-selectively, whereas during nutrient-rich conditions, autophagy selectively eliminates specific cytoplasmic cargo. Depending upon the loaded and digested cytoplasmic cargo, autophagy has been divided into aggrephagy, mitophagy, pexophagy, ER-phagy, xenophagy, and so on [25]. Recently, an alternative pathway of LD degradation through the lysosomal pathway of autophagy has been described and termed lipophagy [26].
- Lipophagy was originally described in hepatocytes, where it is critical for maintaining cellular energy homeostasis in obesity and metabolic syndrome. In vitro and in vivo studies have demonstrated the selective uptake of LDs by autophagosomes, and the genetic or chemical inhibition of autophagy has been shown to reduce the β-oxidation of free fatty acids due to the increased accumulation of lipids and LDs [23]. Importantly, Singh and Cuervo [23] found that impaired autophagy in cultured hepatocytes and mouse liver led to abnormally high levels of hepatic cholesterol along with aberrant triacylglycerol deposition because of the defective clearance of LDs. Furthermore, that study identified a previously unknown function for autophagy in lipid metabolism, with possible implications for various human diseases involving lipid over-accumulation, such as atherosclerosis and cardiovascular disease. Indeed, another report demonstrated that lipophagy became dysfunctional in atherogenesis, and that its deficiency promoted atherosclerosis in part through inflammasome hyperactivation caused by cholesterol crystal accumulation in macrophages [27]. Additionally, macrophage-specific autophagy deficiency led to increased apoptosis and oxidative stress in advanced lesional macrophages, promoted plaque necrosis, and worsened lesional efferocytosis [28]. Thus, the autophagy pathway may contribute to regulate access to lipid stimulation in macrophages in atherosclerotic plaques.
- Consistent with these findings, LAL has been the focus of new studies. LAL is the central enzyme for hydrolysis in lysosomes, and its deficiency leads to human cholesterol storage disorders such as CE storage disease (CESD) and Wolman disease. Bowden et al. [29] provided an explanation of the hypolipoproteinemia seen in CESD patients by showing that LAL activity contributed to the regulation of ATP-binding cassette subfamily A member 1 (ABCA1) expression and activity. Additional studies will be required to clarify the mechanism of LAL, but it is clear that LAL plays an essential role in atherosclerosis via acid lipolysis.
MACROPHAGE CHOLESTEROL EFFLUX
- This macrophage cholesterol efflux function is predominantly mediated by high density lipoprotein (HDL). Apolipoprotein A1 (apoA1), the most abundant protein in HDL, and mature HDL particles serve as acceptors of macrophage cholesterol efflux. Lipid-poor apoA1 promotes the efflux of cholesterol from macrophages via ABCA1, and mature HDL promotes macrophage cholesterol efflux through the ATP-binding cassette subfamily G member 1 (ABCG1) transporter. Additionally mature HDL can directly deliver cholesterol to the liver via scavenger receptor class B member 1 (SR-BI), or indirectly through transfer of cholesterol to apoB-containing lipoproteins, with sequential uptake by LDL receptors in the liver [30]. Both in vitro and in vivo analyses have shown that ATP-binding cassette (ABC) transporter deficiency promotes atherosclerotic lesion development through impaired cholesterol efflux in macrophages [31323334]. Consistent with these findings, lipophagy-mediated macrophage efflux is primarily ABCA1-dependent, since cholesterol delivery to ABCA1 in the lipophagy-defective macrophages is limited by liver X receptor (LXR) [21].
- LXRs are members of the nuclear receptor family of ligand-dependent transcription factors that mediate the regulation of cholesterol homeostasis [35]. LXRs form heterodimers with retinoid X receptors (RXRs) and activate transcription of specific target genes such as those coding for ABC transporters and apoE, which can function as an acceptor; thus, promoting cholesterol transport by the ABCA-1 dependent pathway [3637]. Additionally, LXRs induce the synthesis of fatty acids, which act as substrates of ACAT1 in cholesterol esterification reactions [38]. Despite these results, the relationship of autophagy with LXR and miR-33 [39], which regulates the expression of ABCA1, has yet to be clarified. Therefore, it will be necessary to elucidate the association of LXR and autophagy in macrophage cholesterol homeostasis.
MACROPHAGE CHOLESTEROL EFFLUX
- Macrophages are essential cells that regulate lipid metabolism, especially through RCT, affecting both the progression and regression of atherosclerosis. Therefore, many studies have been conducted to establish a therapeutic strategy for atherosclerosis by studying the cholesterol pathway of macrophages. Recent studies have identified the role of lipophagy, which regulates the acidic hydrolysis of cholesterol from macrophage LDs via the lysosomes, in macrophage RCT and atherosclerosis. Understanding the effective removal of cholesterol from macrophage foam cells, in combination with future studies elucidating the role of lipophagy in macrophages, will lead to the development of novel therapeutic avenues for atherogenesis and metabolic syndrome.
CONCLUSIONS
-
Acknowledgements
- This work was supported by a National Research Foundation of Korea (NRF) grant funded by the Korean government (No. 2012R1A3A2026454 and 2012R1A6A3A04040206).
ACKNOWLEDGMENTS
-
CONFLICTS OF INTEREST: No potential conflict of interest relevant to this article was reported.
Article information
- 1. Ross R. Atherosclerosis: an inflammatory disease. N Engl J Med 1999;340:115–126. ArticlePubMed
- 2. Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation 2002;105:1135–1143. ArticlePubMed
- 3. Mestas J, Ley K. Monocyte-endothelial cell interactions in the development of atherosclerosis. Trends Cardiovasc Med 2008;18:228–232. ArticlePubMedPMC
- 4. Brown MS, Goldstein JL. A proteolytic pathway that controls the cholesterol content of membranes, cells, and blood. Proc Natl Acad Sci U S A 1999;96:11041–11048. ArticlePubMedPMC
- 5. Moore KJ, Freeman MW. Scavenger receptors in atherosclerosis: beyond lipid uptake. Arterioscler Thromb Vasc Biol 2006;26:1702–1711. ArticlePubMed
- 6. de Villiers WJ, Smart EJ. Macrophage scavenger receptors and foam cell formation. J Leukoc Biol 1999;66:740–746. ArticlePubMed
- 7. Kunjathoor VV, Febbraio M, Podrez EA, Moore KJ, Andersson L, Koehn S, et al. Scavenger receptors class A-I/II and CD36 are the principal receptors responsible for the uptake of modified low density lipoprotein leading to lipid loading in macrophages. J Biol Chem 2002;277:49982–49988. ArticlePubMed
- 8. Moore KJ, Kunjathoor VV, Koehn SL, Manning JJ, Tseng AA, Silver JM, et al. Loss of receptor-mediated lipid uptake via scavenger receptor A or CD36 pathways does not ameliorate atherosclerosis in hyperlipidemic mice. J Clin Invest 2005;115:2192–2201. ArticlePubMedPMC
- 9. Kuchibhotla S, Vanegas D, Kennedy DJ, Guy E, Nimako G, Morton RE, et al. Absence of CD36 protects against atherosclerosis in ApoE knock-out mice with no additional protection provided by absence of scavenger receptor A I/II. Cardiovasc Res 2008;78:185–196. ArticlePubMedPDF
- 10. Greaves DR, Gordon S. The macrophage scavenger receptor at 30 years of age: current knowledge and future challenges. J Lipid Res 2009;50:S282–S286. ArticlePubMedPMC
- 11. Febbraio M, Guy E, Silverstein RL. Stem cell transplantation reveals that absence of macrophage CD36 is protective against atherosclerosis. Arterioscler Thromb Vasc Biol 2004;24:2333–2338. ArticlePubMed
- 12. Yagyu H, Kitamine T, Osuga J, Tozawa R, Chen Z, Kaji Y, et al. Absence of ACAT-1 attenuates atherosclerosis but causes dry eye and cutaneous xanthomatosis in mice with congenital hyperlipidemia. J Biol Chem 2000;275:21324–21330. ArticlePubMed
- 13. Fazio S, Major AS, Swift LL, Gleaves LA, Accad M, Linton MF, et al. Increased atherosclerosis in LDL receptor-null mice lacking ACAT1 in macrophages. J Clin Invest 2001;107:163–171. ArticlePubMedPMC
- 14. Nissen SE, Tuzcu EM, Brewer HB, Sipahi I, Nicholls SJ, Ganz P, et al. Effect of ACAT inhibition on the progression of coronary atherosclerosis. N Engl J Med 2006;354:1253–1263. ArticlePubMed
- 15. Yeaman SJ. Hormone-sensitive lipase: new roles for an old enzyme. Biochem J 2004;379(Pt 1):11–22. ArticlePubMedPMCPDF
- 16. Zhao B, Fisher BJ, St Clair RW, Rudel LL, Ghosh S. Redistribution of macrophage cholesteryl ester hydrolase from cytoplasm to lipid droplets upon lipid loading. J Lipid Res 2005;46:2114–2121. ArticlePubMed
- 17. Sekiya M, Osuga J, Nagashima S, Ohshiro T, Igarashi M, Okazaki H, et al. Ablation of neutral cholesterol ester hydrolase 1 accelerates atherosclerosis. Cell Metab 2009;10:219–228. ArticlePubMed
- 18. Igarashi M, Osuga J, U ozaki H, Sekiya M, Nagashima S, Takahashi M, et al. The critical role of neutral cholesterol ester hydrolase 1 in cholesterol removal from human macrophages. Circ Res 2010;107:1387–1395. ArticlePubMed
- 19. Ouimet M, Marcel YL. Regulation of lipid droplet cholesterol efflux from macrophage foam cells. Arterioscler Thromb Vasc Biol 2012;32:575–581. ArticlePubMed
- 20. Avart SJ, Bernard DW, Jerome WG, Glick JM. Cholesteryl ester hydrolysis in J774 macrophages occurs in the cytoplasm and lysosomes. J Lipid Res 1999;40:405–414. ArticlePubMed
- 21. Ouimet M, Franklin V, Mak E, Liao X, Tabas I, Marcel YL. Autophagy regulates cholesterol efflux from macrophage foam cells via lysosomal acid lipase. Cell Metab 2011;13:655–667. ArticlePubMedPMC
- 22. Kundu M, Thompson CB. Autophagy: basic principles and relevance to disease. Annu Rev Pathol 2008;3:427–455. ArticlePubMed
- 23. Singh R, Cuervo AM. Autophagy in the cellular energetic balance. Cell Metab 2011;13:495–504. ArticlePubMedPMC
- 24. Swanson MS, Byrne BG, Dubuisson JF. Kinetic analysis of autophagosome formation and turnover in primary mouse macrophages. Methods Enzymol 2009;452:383–402. ArticlePubMed
- 25. Jin M, Liu X, Klionsky DJ. SnapShot: selective autophagy. Cell 2013;152:368–368.e2. ArticlePubMedPMC
- 26. Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, et al. Autophagy regulates lipid metabolism. Nature 2009;458:1131–1135. ArticlePubMedPMCPDF
- 27. Razani B, Feng C, Coleman T, Emanuel R, Wen H, Hwang S, et al. Autophagy links inflammasomes to atherosclerotic progression. Cell Metab 2012;15:534–544. ArticlePubMedPMC
- 28. Liao X, Sluimer JC, Wang Y, Subramanian M, Brown K, Pattison JS, et al. Macrophage autophagy plays a protective role in advanced atherosclerosis. Cell Metab 2012;15:545–553. ArticlePubMedPMC
- 29. Bowden KL, Bilbey NJ, Bilawchuk LM, Boadu E, Sidhu R, Ory DS, et al. Lysosomal acid lipase deficiency impairs regulation of ABCA1 gene and formation of high density lipoproteins in cholesteryl ester storage disease. J Biol Chem 2011;286:30624–30635. ArticlePubMedPMC
- 30. Rader DJ, Alexander ET, Weibel GL, Billheimer J, Rothblat GH. The role of reverse cholesterol transport in animals and humans and relationship to atherosclerosis. J Lipid Res 2009;50:S189–S194. ArticlePubMedPMC
- 31. van Eck M, Bos IS, Kaminski WE, Orso E, Rothe G, Twisk J, et al. Leukocyte ABCA1 controls susceptibility to atherosclerosis and macrophage recruitment into tissues. Proc Natl Acad Sci U S A 2002;99:6298–6303. ArticlePubMedPMC
- 32. Clee SM, Zwinderman AH, Engert JC, Zwarts KY, Molhuizen HO, Roomp K, et al. Common genetic variation in ABCA1 is associated with altered lipoprotein levels and a modified risk for coronary artery disease. Circulation 2001;103:1198–1205. ArticlePubMed
- 33. Wang X, Collins HL, Ranalletta M, Fuki IV, Billheimer JT, Rothblat GH, et al. Macrophage ABCA1 and ABCG1, but not SR-BI, promote macrophage reverse cholesterol transport in vivo. J Clin Invest 2007;117:2216–2224. ArticlePubMedPMC
- 34. Kennedy MA, Barrera GC, Nakamura K, Baldan A, Tarr P, Fishbein MC, et al. ABCG1 has a critical role in mediating cholesterol efflux to HDL and preventing cellular lipid accumulation. Cell Metab 2005;1:121–131. ArticlePubMed
- 35. Beaven SW, Tontonoz P. Nuclear receptors in lipid metabolism: targeting the heart of dyslipidemia. Annu Rev Med 2006;57:313–329. ArticlePubMed
- 36. Chawla A, Repa JJ, Evans RM, Mangelsdorf DJ. Nuclear receptors and lipid physiology: opening the X-files. Science 2001;294:1866–1870. ArticlePubMed
- 37. Laffitte BA, Repa JJ, Joseph SB, Wilpitz DC, Kast HR, Mangelsdorf DJ, et al. LXRs control lipid-inducible expression of the apolipoprotein E gene in macrophages and adipocytes. Proc Natl Acad Sci U S A 2001;98:507–512. ArticlePubMedPMC
- 38. Repa JJ, Liang G, Ou J, Bashmakov Y, Lobaccaro JM, Shimomura I, et al. Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRalpha and LXRbeta. Genes Dev 2000;14:2819–2830. ArticlePubMedPMC
- 39. Rayner KJ, Suarez Y, Davalos A, Parathath S, Fitzgerald ML, Tamehiro N, et al. MiR-33 contributes to the regulation of cholesterol homeostasis. Science 2010;328:1570–1573. ArticlePubMedPMC
References
Fig. 1
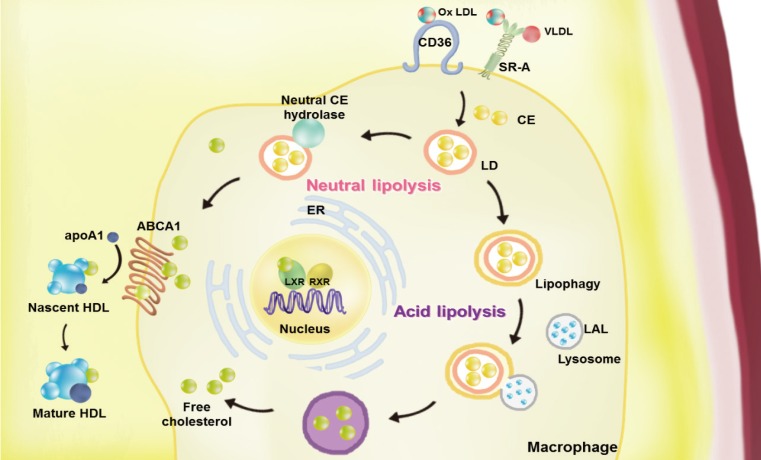
Overview of the pathways of macrophage lipoprotein uptake and efflux. Macrophages uptake very low density lipoprotein (VLDL) and modified low density lipoprotein (LDL), such as oxidized (Ox) LDL via scavenger receptors (SRs, including by SR-A1 and cluster of differentiation 36 [CD36]). The internalized LDL is esterified by acetyl-coenzyme A acetyltransferases (ACAT1) and stored in lipid droplets (LDs). Neutral and acid lipolysis contribute to the release of cholesteryl ester (CE) for efflux in LDs via neutral CE hydrolase or lipophagy through lysosomal acid lipase (LAL). The cellular free cholesterol activates the liver X receptor (LXR)-retinoid X receptor (RXR) heterodimeric transcription factor that upregulates expression of ATP-binding cassette subfamily A member 1 (ABCA1). This transporter mediates the free cholesterol efflux from macrophages, with lipid-poor apolipoprotein A1 (apoA1) used as an acceptor. By reducing the accumulation of cholesterol in the wall of arteries via macrophage cholesterol efflux, reverse cholesterol transport may the prevent development of atherosclerosis. ER, endoplasmic reticulum; HDL, high density lipoprotein.
Figure & Data
References
Citations
Citations to this article as recorded by 
- Bile salt signaling and bile salt-based therapies in cardiometabolic disease
Claire C.J. Groenen, Thuc-Anh Nguyen, Coen C. Paulusma, Stan F.J. van de Graaf
Clinical Science.2024; 138(1): 1. CrossRef - Huazhuotongmai decoction exerts anti-atherosclerotic effects by modulating the expression of ABCA1/SR-B1/PPAR-γ in vivo and in vitro
Ya-ru Yan, Zi-jun Jia, Ya Wang, Feng-qin Xu, Qing-bing Zhou
Phytomedicine Plus.2023; 3(2): 100436. CrossRef - The SGLT2 Inhibitor Canagliflozin Reduces Atherosclerosis by Enhancing Macrophage Autophagy
Hongping Chen, Da Teng, Bowen Xu, Chunxiao Wang, Hua Wang, Wenjuan Jia, Lei Gong, Haibin Dong, Lin Zhong, Jun Yang
Journal of Cardiovascular Translational Research.2023; 16(5): 999. CrossRef - FURIN suppresses the progression of atherosclerosis by promoting macrophage autophagy
Hongping Chen, Lihui Zhang, Shaohua Mi, Hua Wang, Chunxiao Wang, Wenjuan Jia, Lei Gong, Haibin Dong, Bowen Xu, Yanyan Jing, Peipei Ge, Zhigang Pei, Lin Zhong, Jun Yang
The FASEB Journal.2023;[Epub] CrossRef - Unbalanced Redox With Autophagy in Cardiovascular Disease
Se-Jin Jeong, Goo Taeg Oh
Journal of Lipid and Atherosclerosis.2023; 12(2): 132. CrossRef - Edible Bird’s Nest Effectively Attenuates Atherosclerosis through Modulation of Cholesterol Metabolism via Activation of PPARγ/LXRα Signaling Pathway In Vivo
Nurul Nadiah Mohamad Nasir, Ramlah Mohamad Ibrahim, Rozi Mahmud, Nor Asma Ab Razak, Norsharina Ismail, Kim Wei Chan, Md Zuki Abu Bakar, Akhilesh K. Verma
Journal of Food Biochemistry.2023; 2023: 1. CrossRef - The mitochondrial translocator protein (TSPO, 18 kDa): A key multifunctional molecule in liver diseases
Yuchang Li, Liting Chen, Vassilios Papadopoulos
Biochimie.2023;[Epub] CrossRef - The Differential Metabolomes in Cumulus and Mural Granulosa Cells from Human Preovulatory Follicles
Er-Meng Gao, Bongkoch Turathum, Ling Wang, Di Zhang, Yu-Bing Liu, Rong-Xin Tang, Ri-Cheng Chian
Reproductive Sciences.2022; 29(4): 1343. CrossRef - PHLPP1 promotes neutral lipid accumulation through AMPK/ChREBP-dependent lipid uptake and fatty acid synthesis pathways
Keerthana Balamurugan, Raghavender Medishetti, Jyothi Kotha, Parameshwar Behera, Kanika Chandra, Vijay Aditya Mavuduru, Manjunath B. Joshi, Ramesh Samineni, Madhumohan R. Katika, Writoban Basu Ball, Manjunatha Thondamal, Anil Challa, Kiranam Chatti, Kisho
iScience.2022; 25(2): 103766. CrossRef - Hydrochloride Berberine ameliorates alcohol-induced liver injury by regulating inflammation and lipid metabolism
Xiumei Ke, Ruoyu Zhang, Pan Li, Ling Zuo, Meng Wang, Junxuan Yang, Jianwei Wang
Biochemical and Biophysical Research Communications.2022; 610: 49. CrossRef - Metabolic Regulation of Macrophage Activation
Ourania Kolliniati, Eleftheria Ieronymaki, Eleni Vergadi, Christos Tsatsanis
Journal of Innate Immunity.2022; 14(1): 51. CrossRef - Emerging Roles of Lipophagy in Cancer Metastasis
Haimeng Yin, Ying Shan, Tian Xia, Yan Ji, Ling Yuan, Yiwen You, Bo You
Cancers.2022; 14(18): 4526. CrossRef - Genetic Factors for Coronary Heart Disease and Their Mechanisms: A Meta-Analysis and Comprehensive Review of Common Variants from Genome-Wide Association Studies
Khairul Anwar Zarkasi, Noraidatulakma Abdullah, Nor Azian Abdul Murad, Norfazilah Ahmad, Rahman Jamal
Diagnostics.2022; 12(10): 2561. CrossRef - Caveolin-1 in autophagy: A potential therapeutic target in atherosclerosis
Kai Hou, Shuai Li, Meng Zhang, Xuping Qin
Clinica Chimica Acta.2021; 513: 25. CrossRef - Contradictory regulation of macrophages on atherosclerosis based on polarization, death and autophagy
Jing Zhang, Chuan-Rui Ma, Yun-Qing Hua, Lan Li, Jing-Yu Ni, Yu-Ting Huang, Sophia Esi Duncan, Sheng Li, Shan Gao, Guan-Wei Fan
Life Sciences.2021; 276: 118957. CrossRef - A meta-analysis of HDL cholesterol efflux capacity and concentration in patients with rheumatoid arthritis
Binbin Xie, Jiang He, Yong Liu, Ting Liu, Chaoqun Liu
Lipids in Health and Disease.2021;[Epub] CrossRef - Rosmarinic Acid Increases Macrophage Cholesterol Efflux through Regulation of ABCA1 and ABCG1 in Different Mechanisms
Jean-Baptiste Nyandwi, Young Shin Ko, Hana Jin, Seung Pil Yun, Sang Won Park, Hye Jung Kim
International Journal of Molecular Sciences.2021; 22(16): 8791. CrossRef - FGF21 induces autophagy‐mediated cholesterol efflux to inhibit atherogenesis via RACK1 up‐regulation
Lin Xiaolong, Guo Dongmin, Mihua Liu, Wang Zuo, Hu Huijun, Tan Qiufen, Hu XueMei, Lin Wensheng, Pan Yuping, Lin Jun, Zeng Zhaolin
Journal of Cellular and Molecular Medicine.2020; 24(9): 4992. CrossRef - Lipophagy in atherosclerosis
Qing Liu, Yuan-Mei Wang, Hong-Feng Gu
Clinica Chimica Acta.2020; 511: 208. CrossRef - Lysosomotropic Features and Autophagy Modulators among Medical Drugs: Evaluation of Their Role in Pathologies
Tatiana A. Korolenko, Thomas P. Johnston, Vaclav Vetvicka
Molecules.2020; 25(21): 5052. CrossRef - LncRNA MALAT1 Enhances ox-LDL-Induced Autophagy through the SIRT1/MAPK/NF-κB Pathway in Macrophages
Jiaqi Yang, Xuze Lin , Liangshan Wang, Tienan Sun, Qi Zhao, Qian Ma, Yujie Zhou
Current Vascular Pharmacology.2020; 18(6): 652. CrossRef -
CTRP13 inhibits atherosclerosis
via
autophagy‐lysosome‐dependent degradation of CD36
Cheng Wang, Wenjing Xu, Minglu Liang, Dan Huang, Kai Huang
The FASEB Journal.2019; 33(2): 2290. CrossRef - Subclinical atherosclerosis and its progression are modulated by PLIN2 through a feed‐forward loop between LXR and autophagy
P. Saliba‐Gustafsson, M. Pedrelli, K. Gertow, O. Werngren, V. Janas, S. Pourteymour, D. Baldassarre, E. Tremoli, F. Veglia, R. Rauramaa, A.J. Smit, P. Giral, S. Kurl, M. Pirro, U. de Faire, S.E. Humphries, A. Hamsten, I. Gonçalves, M. Orho‐Melander, A. Fr
Journal of Internal Medicine.2019; 286(6): 660. CrossRef - PCSK9: A new participant in lipophagy in regulating atherosclerosis?
Jun Xiao, Yi-Min Deng, Xiang-Rui Liu, Jian-Ping Cao, Min Zhou, Ya-Ling Tang, Wen-Hao Xiong, Zhi-Sheng Jiang, Zhi-Han Tang, Lu-Shan Liu
Clinica Chimica Acta.2019; 495: 358. CrossRef - Autophagy-Mediated Cholesterol Trafficking Controls Steroid Production
Michael J. Texada, Alina Malita, Christian F. Christensen, Kathrine B. Dall, Nils J. Faergeman, Stanislav Nagy, Kenneth A. Halberg, Kim Rewitz
Developmental Cell.2019; 48(5): 659. CrossRef - Lipophagy in nonliver tissues and some related diseases: Pathogenic and therapeutic implications
Kebing Zhou, Pingbo Yao, Jun He, Hong Zhao
Journal of Cellular Physiology.2019; 234(6): 7938. CrossRef - Autophagy differentially regulates macrophage lipid handling depending on the lipid substrate (oleic acid vs. acetylated-LDL) and inflammatory activation state
Sapir Hadadi-Bechor, Yulia Haim, Tal Pecht, Roni Gat, Tanya Tarnovscki, Martin Gericke, Assaf Rudich
Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids.2019; 1864(12): 158527. CrossRef - Autophagy and Age-Related Eye Diseases
Xue Yang, Xinan Pan, Xiaorui Zhao, Jin Luo, Mingpu Xu, Daoming Bai, Yan Hu, Xu Liu, Qiongfang Yu, Dian Gao
BioMed Research International.2019; 2019: 1. CrossRef - Foam cell formation and cholesterol trafficking and metabolism disturbances in atherosclerosis
Alexandrina Volobueva, Dongwei Zhang, Andrey V. Grechko, Alexander N. Orekhov
Cor et Vasa.2019; 61(1): 48. CrossRef - Oxidative Stress, Lipid Peroxidation, and Loss of Hyaluronic Acid in the Human Vitreous Affected by Synchysis Scintillans
Loredana Bergandi, Oleksii A Skorokhod, Rosalba La Grotta, Evelin Schwarzer, Raffaele Nuzzi
Journal of Ophthalmology.2019; 2019: 1. CrossRef - Programmed cell death protein 4 deficiency suppresses foam cell formation by activating autophagy in advanced glycation end‐product low‐density lipoprotein–induced macrophages
Shan Li, Guangdong Gao, Fuyun Wu, Dan Liu, Hongyan Zhao, Jing Ke, Ying Liu, Fei Li, Jian Li, Zongyun Chen, Zhiming Tang, Lei Bai, Jinxuan Zhang, Wei Zheng, Xin Chen
Journal of Cellular Biochemistry.2019; 120(5): 7689. CrossRef - Mindin deficiency in macrophages protects against foam cell formation and atherosclerosis by targeting LXR-β
Cheng Zhang, Juan-Juan Qin, Fu-Han Gong, Jing-Jing Tong, Wen-Lin Cheng, Haiping Wang, Yan Zhang, Xueyong Zhu, Zhi-Gang She, Hao Xia, Li-Hua Zhu
Clinical Science.2018; 132(11): 1199. CrossRef - LJ-1888, a selective antagonist for the A3 adenosine receptor, ameliorates the development of atherosclerosis and hypercholesterolemia in apolipoprotein E knock-out mice
Jong-Gil Park, Se-Jin Jeong, Jinha Yu, Gyudong Kim, Lak Shin Jeong, Goo Taeg Oh
BMB Reports.2018; 51(10): 520. CrossRef - Intracellular and Plasma Membrane Events in Cholesterol Transport and Homeostasis
Dmitry Y. Litvinov, Eugeny V. Savushkin, Alexander D. Dergunov
Journal of Lipids.2018; 2018: 1. CrossRef
 PubReader
PubReader Cite
Cite