Novel Mutation in PTHLH Related to Brachydactyly Type E2 Initially Confused with Unclassical Pseudopseudohypoparathyroidism
Article information

Abstract
Background
Autosomal-dominant brachydactyly type E is a congenital abnormality characterized by small hands and feet, which is a consequence of shortened metacarpals and metatarsals. We recently encountered a young gentleman exhibiting shortening of 4th and 5th fingers and toes. Initially, we suspected him having pseudopseudohypoparathyroidism (PPHP) because of normal biochemical parameters, including electrolyte, Ca, P, and parathyroid hormone (PTH) levels; however, his mother and maternal grandmother had the same conditions in their hands and feet. Furthermore, his mother showed normal biochemical parameters. To the best of our knowledge, PPHP is inherited via a mutated paternal allele, owing to the paternal imprinting of GNAS (guanine nucleotide binding protein, alpha stimulating) in the renal proximal tubule. Therefore, we decided to further analyze the genetic background in this family.
Methods
Whole exome sequencing was performed using genomic DNA from the affected mother, son, and the unaffected father as a negative control.
Results
We selected the intersection between 45,490 variants from the mother and 45,646 variants from the son and excluded 27,512 overlapping variants identified from the father. By excluding homogenous and compound heterozygous variants and removing all previously reported variants, 147 variants were identified to be shared by the mother and son. Variants that had least proximities among species were excluded and finally 23 variants remained.
Conclusion
Among them, we identified a defect in parathyroid hormone like hormone (PTHLH), encoding the PTH-related protein, to be disease-causative. Herein, we report a family affected with brachydactyly type E2 caused by a novel PTHLH mutation, which was confused with PPHP with unclassical genetic penetrance.
INTRODUCTION
Brachydactyly (BD) is a phenotype characterized by short fingers and toes, and is classified into five types, A to E, based on anatomical and genetic traits [1]. Among them, BD type E (BDE) is characterized by the variable shortening of metacarpals/metatarsals and frequent involvement of phalanges [2]. BDE may occur as an isolated trait or part of several syndromes related to multihormone resistance, acrodysostosis, hypertension, short stature, mental retardation, and Turner syndrome [3]. BDE has been classified into three distinct varieties: type E1 (OMIM 113300) caused by HOXD13 (homeobox D13) mutations, with shortening of metacarpal IV, sometimes associated with shortening of metatarsal IV; type E2 (OMIM 613382) caused by parathyroid hormone like hormone (PTHLH) mutations, with shortening of metacarpals IV and V associated with shortening of the distal phalanx of the thumb; and type E3, with various combinations of short metacarpals [34]. Pseudohypoparathyroidism (PHP), characterized by resistance to parathyroid hormone (PTH), is a commonly observed disorder related to hormone resistance [5]. To this day, BDE is observed in 70% to 78% of patients with PHP [6]. The clinical manifestations of PHP are diverse, including short stature, round face, obesity, intramembranous calcifications, BD, and intellectual deficiency; these clinical features characterize Albright's hereditary osteodystrophy (AHO). Because of PTH resistance, individuals with PHP develop hypocalcemia and hyperphosphatemia [57]. However, pseudopseudohypoparathyroidism (PPHP) is described as a condition with the features of AHO, yet without PTH resistance. Tissue-specific haploinsufficiency and genomic imprinting form the basis for the similarities and differences between the clinical features of PHP Ia and PPHP [8]. Haploinsufficiency of Gsα, which is the α subunit of the Gs protein, could be the reason for the occurrence of AHO features in PHP Ia and PPHP [9]. One normal Gsα allele is not sufficient for a normal Gsα function, thereby resulting in the development of AHO features. These are the results of reduced chondrocyte proliferation and early chondrocyte differentiation, caused by partial resistance to PTH-related peptide (PTHrP) [10]. Tissue-specific Gsα imprinting gives rise to the different features distinguishing between PHP Ia and PPHP, such as multihormone resistance [11]. In certain tissues, wherein the maternal allele is preferentially expressed, and if mutations occur in the maternal allele, there will be no or extremely limited expression Gsα. Mutations in the active maternal allele result in severe Gsα deficiency and hormone resistance, collectively termed PHP Ia, and development of resistance to multiple Gs protein-coupled hormones (e.g., thyroid-stimulating hormone, growth hormone-releasing hormone, and PTH) [12131415]. However, inherited mutations in the relatively inactive paternal allele have extremely limited effect on Gsα expression or hormone action. Mutations in the paternal allele result in developmental defects of AHO without hormonal resistance, termed PPHP [1116].
Herein, we report the cases of a patient and his mother, who presented with BDE2, initially confused for unclassical PPHP, and a novel BDE-related mutation in PTHLH identified by exome sequencing.
METHODS
Ethics statement
The study was approved by the Institutional Review Board (IRB) of Gachon University Gil Medical Center, Korea (IRB No. GIRBA2151). Informed consent was obtained from all subjects before participation and all research was performed in accordance with relevant guidelines and regulations.
Clinical report
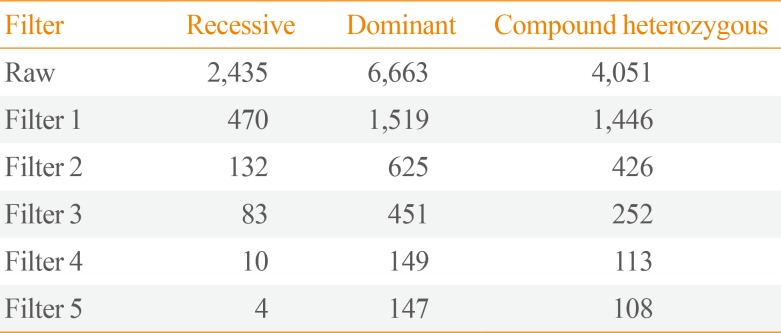
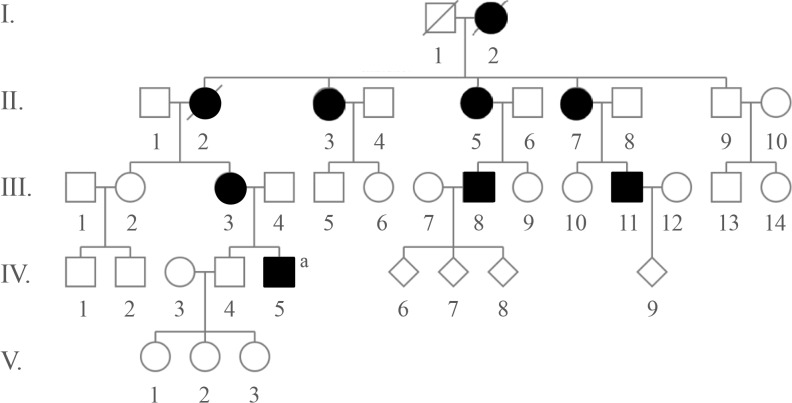
A 31-year-old Caucasian gentleman (IV-5) (Fig. 1) visited the clinic with visibly short fingers and toes on both sides, especially the 4th and 5th (Fig. 2A, C). His height was 175 cm, and weight was 60 kg. Upon physical examination, no typical abnormality, such as round face, short neck, nor short stature was revealed, and no specific complaints were noted. Cervical inspection and palpation showed that the thyroid gland was not enlarged. Laboratory tests, including complete blood count and urine analysis, were normal. All values obtained during the serum chemistry test were within normal limits. Additionally, calcium and phosphorous levels in both the serum and 24-hour urine sample were within normal limits. Serum PTH level and thyroid function tests were normal. He reported that his 66-year-old mother (III-3) (Fig. 1) and deceased maternal grandmother (II-2) (Fig. 1) showed almost the same phenotype in their hands and feet, which was initially considered to be AHO (Fig. 2B, D). The patient's mother did not have short stature; breast development was normal, and there was no problem with breastfeeding. On a simple X-ray, the hands and feet of the patient showed shortening of the 4th and 5th metacarpals and 4th and 5th metatarsals. Premature fusion of the epiphyses was also observed in the shortened bones (Fig. 2E). The proband and his mother were initially considered to have PPHP, as serum calcium, phosphorus, and PTH levels were normal. However, this was inconsistent with our knowledge that in PPHP, paternal silencing of GNAS (guanine nucleotide binding protein, alpha stimulating) in the renal proximal tubule occurs. Consequently, AHO is inherited from the father, which appeared to be quite contrary in this pedigree (Fig. 1).
Pedigree of this family. Brachydatylies are found in every generations in both genders indicating autosomal dominant trait of inheritance. Open, unaffected; closed, affected. aProband.
Clinical phenotypes of the proband and his mother: (A) right hand of the proband; (B) left hand of the mother; (C) right foot of the proband; (D) left foot of the mother, note the shortening of the 4th and 5th metacarpals and 4th and 5th metatarsals; (E) radiological feature of the mother's left feet showing shortening of 4th and 5th metatarsals. In addition, premature fusion of the epiphyses is noted in the shortened bones.
Whole exome sequencing
Whole exome sequencing was performed using genomic DNA of the patients (proband and his mother) and his father (III-4) (Fig. 1) as the negative control at the Theragen Etex Bio Institute (Suwon, Korea). The selected genomic DNA sample was randomly fragmented using Covaris (Covaris Inc., Woburn, MA, USA), and the size of the library fragments ranged mainly between 150 and 200 bp. Adapters were then ligated to both ends of the fragments. The adapter-ligated templates were purified using Agencourt AMPure (Beckman Coulter, Beverly, MA, USA) SPRI (solid-phase reversible immobilization) beads, and fragments with an insert size of approximately 250 bp were excised. Extracted DNA was amplified by ligation-mediated polymerase chain reaction (LM-PCR), purified, and hybridized to the Sure-Select biotinylated RNA Library (baits) for enrichment. The hybridized fragments were bound to streptavidin beads, while nonhybridized fragments were washed out after 24 hours. Captured LM-PCR products were subjected to the estimation of the enrichment magnitude using an Agilent 2100 Bioanalyzer (Agilent Genomics, Santa Clara, CA, USA). Each captured library was then loaded onto a Hiseq 2000 platform (Illumina, San Diego, CA, USA), and high-throughput sequencing was performed for each captured library to ensure that each sample had the desired average sequencing depth. Raw image files were processed by HiSeq Control Software (HCS) 1.4.8 for base-calling with default parameters, and the sequences of each individual were generated as 90-bp paired-end reads.
Raw data filtering
Generated reads have been filtered with sickle (v.1.33) and aligned to the hg19 using Burrows Wheeler Aligner (v.0.7.12). Aligned reads were processed and duplicate read were removed using PicardTools (v.1.98). Variant calling and quality filtration was performed using GATK best practices variant quality score recalibration (v.2.3-9). Single-nucleotide polymorphism and indel functionally annotation was performed using SnpEff (v.4.1) and frequency annotation was performed using 1000 Genome Project (phase3), EXAC, ESP6500 and In-house database. In the next step, variants in patient samples were filtered gradationally as follows: In filter 1 (Region Filter), the variants included missense, nonsense, loss-of-stop codon, insertion/deletion, and splicesite mutations. It annotated and predicted the effects of variants on genes (such as amino acid changes) with SnpEff (v.4.1). In filter 2 (SIFT and PolyPhen-2 Filters), dbNSFP (version 2.9) is a database developed for functional prediction and annotation of all potential non-synonymous single-nucleotide variants (ns-SNVs) in the human genome [17]. With the SIFT and Polyphen-2 HDIV scores, it predicted whether an amino acid substitution affected protein function. It was expressed as “D (damaging),” and “T (tolerated)” in the SIFT score, and “possibly damaging,” “benign,” “neutral,” and “deleterious” in the Polyphen-2 HDIV score. We removed the variants which are predicted “T” in the SIFT score and “benign” in PolyPhen-2 HDIV score. In filter 3 (PhastCons Filter), the PhastCons score measures the strength of purifying selection acting on a DNA sequence. A high PhastCons score (0.2) may be considered strong evidence for the functional importance of a genomic region. The variants had PhastCons score greater than 0.2 from PhastCons 100 way. In filter 4 (1000 Genomes Filter), among 1000 Genome allele frequency information of Phage3 1000G db (n=2,504), the (alt allele) variants had a frequency less than 0.01 or unknown. Filter 5 (In-House Filter) selected variants with a frequency less than 0.01 or unknown from In-House DB. Namely, Koreans pick up quite rarelow variation as well.
Genetic analysis
Genomic DNA was extracted from the peripheral blood of all patients using a standard method. All coding exons and intronexon junctions for the genes were amplified and sequenced. PCR was performed using 5 mM MgCl2, 200 µM deoxyribonucleotides, 0.5 µM of each primer, 1 unit of Taq polymerase, and 100 ng of genomic DNA as the template. Primer sequences are available upon request. The PCR products were electrophoresed on polyacrylamide gels, and read in both directions using an ABI PRISM 377 DNA Sequencer (Applied Biosystems, Foster City, CA, USA).
RESULTS
We obtained 45,490 variants from the mother and 45,646 variants from the son. We selected the intersection between the two sets, and filtered out 27,512 overlapping variants identified from the father, who was an unaffected individual (Fig. 3). The remaining 6,663 variants, excluding homogenous and compound heterozygous variants, were then filtered against the dbSNP and In-House databases, removing all previously reported variants. After filtering as described above, totally, 147 variants (139 SNVs and one insertion with seven deletions) were identified to be shared by the two affected individuals (Table 1). All genetic data about the variants are available upon request. Additionally, dbNSFP phyloP46way primate, placental and vertebrate scores were applied. The larger the score, the more conserved the site was in the species. We excluded variants that had a vertebrate, placental, and primate score less than 2, 2.5, and 0.6, respectively. Consequently, 23 variants remained finally (Table 2). Among these, we noted that the c.169C>T mutation in PTHLH encoding PTHrP introduced a stop codon at position 57 (p. Arg57*), and confirmed this mutation by direct targeted gene-sequencing (data not shown).
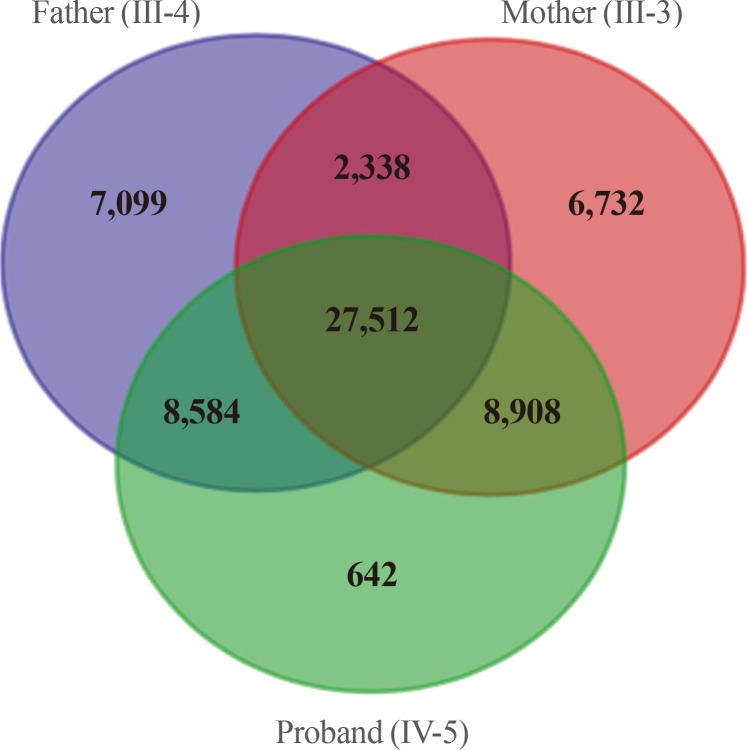
Filters used in analysis of the exome data and numbers of candidate variants. The autosomal dominant variants identified by whole exome sequencing were represented by the Venn diagram. After filtering, a total of 8,908 variants were identified as being shared by the two affected subjects.

Steps of Filtering

Twenty Three Variants Remained after Advanced Steps for the Identification of the Disease-Causing Mutation
DISCUSSION
Thus far, BD has been understood as a type of developmental disorder. When several signaling molecules and pathways involved in developmental processes are disturbed, individuals may consequently present with BD [18]. The PTHrP encoded by PTHLH can be considered a key molecule in BDE, because of its various physiological roles, such as in cartilage development, mammary gland development, lactation, tooth development, expression in the central nervous system, and effects on the smooth muscle [19]. In cartilage development, PTHrP functions to accelerate cartilage cell proliferation, and prevent their progression to terminal proliferation [2021]. PTHrP, acting with other gene products involved in patterning, such as Indian hedgehog protein (IHH), is involved in the proliferation and patterning of bones [22]. Breast development is accomplished by complex epithelial-mesenchymal interactions regulated by essential signaling factors, including PTHrP [23]. During lactation, large amounts of PTHrP, which could be responsible for the adaptation of maternal calcium homeostasis, are produced in glandular epithelial and myoepithelial cells. PTHrP may elevate 1,25-dihydroxyvitamin D and blood calcium levels, and reduce PTH level to maintain maternal calcium metabolism [24]. Teeth formation requires PTHrP, which is secreted by the epithelial layer, and activates alveolar bone resorption by osteoclasts [25]. PTHrP is expressed throughout the central nervous system; it inhibits calcium entry, and protects neurons against glutamate-induced excitotoxicity, resulting from the activation of voltage-dependent calcium channels [26]. The effect of PTHrP on the smooth muscle is the response to stretching by relaxing smooth muscles where PTHrP acts as a smooth-muscle dilator [27].
As mentioned above, the physiological functions of PTHrP are diverse and complex. PTHLH mutations were involved in autosomal-dominant BDE in several reports. Nine intragenic point mutations have been reported [28293031]. We noted the novel c.169C>T mutation in PTHLH which introduced a stop codon at position 57 (p.R57*) (Fig. 4). Multiple physiological dysfunctions induced by those mutations in PTHLH are observed in various clinical phenotypes of BD. Previously reported patients with PTHLH-related BDE had varied BD appearance, short stature, abnormal arm span/height ratio, dental problems, and mental retardation, or did not have some of these features [29]. Our patients had shortening of the 4th and 5th metacarpals and metatarsals, and premature fusion of the epiphyses was observed. However, other abnormalities were not present in our case. Moreover, his mother had no abnormality in height and breast development or with breastfeeding. This also includes no other apparent abnormalities (round face, short neck, or short stature). From the previous reports, there is no conclusive correlation of genotypephenotype in PTHLH mutations related to BDE2 [32]. Interestingly, similar PTHLH mutation, c.166C>T (p.R56*), was reported recently by Jamsheer et al. [31] and by Pereda et al. [32]. This PTHLH mutation is located just beside our reported mutation and the consequences of these mutations are same as introducing premature stop codon. In addition, the clinical phenotypes, such as the normal stature of patient who had c.166C>T (p.R56*) PTHLH mutation are similar to our patients. As described above, PTHLH-related BDE2 could exhibit various clinical features. This makes it difficult to diagnose PTHLH-related BDE2, particularly confusing with PPHP, by clinical features before genetic investigation. The characteristics of patients with PPHP are similar to those exhibited in PTHLH-related BDE2. Short stature, BD, and mental retardation can be observed in both diseases, although hormone resistance cannot. In particular, BD, such as shortening of the fourth and/or fifth metacarpals, and the major clinical signs of AHO were evident in the previous report [33]. In the report, the patients had BD (shortening of the fourth and fifth metacarpals, and metatarsals); however, they did not exhibit PTH resistance (normal calcium and phosphate levels). Other features of commonly seen in AHO, such as short stature, round face, obesity, and intramembranous calcifications and those in PTHLH mutations such as short stature, reduced arm span/height ratio, and dental problems were not observed in these patients. According to these results, it was clinically sufficient to suppose PPHP in the patients. However, PTHLH mutation could be confirmed as a causative factor of BD in these patients by the genetic analysis and PPHP could be excluded.
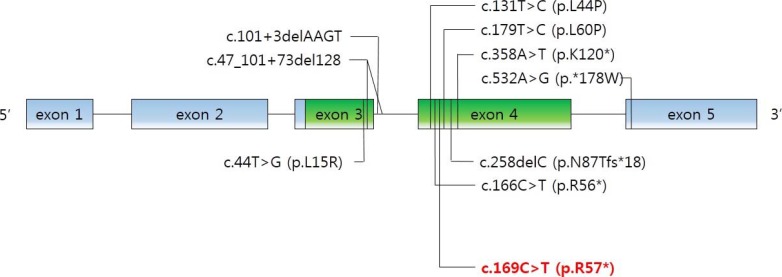
Schematic structure of PTHLH (parathyroid hormone like hormone) gene. Mutations reported in the literature that were associated with brachydactyly type E are indicated in black in addition to the c.169C>T mutation identified in this study in red.
The most commonly used classification system for BD, suggested by Bell [33] and further developed by Temtamy and McKusick [2], is based on anatomical grounds. The various types of BD are confirmed based on clinical phenotypes with inherited traits. The identification of disease-related genes and molecular pathways corresponds to the classification proposed by Bell [18]. However, numerous BD conditions imply that the same BD phenotype could be present in related states, as well as in distinct diseases [34]. Therefore, the identification of the molecular causes of BD should be more advanced, helping us in diagnosing diseases related to BD.
In conclusion, this work reports on a novel PTHLH mutation providing another piece of evidence for PTHLH-related BDE2, and assists us in establishing a differential diagnosis from PPHP.
ACKNOWLEDGMENTS
We thank all family members for their participation. We also thank Drs. Harald Jueppner, Giedre Grigelioniene and Shigeki Nishimori for their insightful advices and Ms. Monica Reyes for technical support. This work was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT and Future Planning, Republic of Korea (NRF-2013R1A1A1A05005629).
Notes
AUTHOR CONTRIBUTIONS: Conception or design: S.L. Acquisition, analysis, or interpretation of data: D.E.L., S.Y.P., S.L. Drafting the work or revising: J.B., H.S.C., S.L. Final approval of the manuscript: S.L.
CONFLICTS OF INTEREST: No potential conflict of interest relevant to this article was reported.