Articles
- Page Path
- HOME > Endocrinol Metab > Volume 33(3); 2018 > Article
-
Review ArticleRecent Topics in Fibrodysplasia Ossificans Progressiva
-
Takenobu Katagiri1,2
 , Sho Tsukamoto1,2, Yutaka Nakachi1, Mai Kuratani1
, Sho Tsukamoto1,2, Yutaka Nakachi1, Mai Kuratani1 -
Endocrinology and Metabolism 2018;33(3):331-338.
DOI: https://doi.org/10.3803/EnM.2018.33.3.331
Published online: September 18, 2018
1Division of Pathophysiology, Research Center for Genomic Medicine, Saitama Medical University, Saitama, Japan.
2Project of Clinical and Basic Research for FOP, Saitama Medical University, Saitama, Japan.
- Corresponding author: Takenobu Katagiri. Division of Pathophysiology, Research Center for Genomic Medicine, Saitama Medical University, 1397-1 Yamane, Hidaka-shi, Saitama 350-1241, Japan. Tel: +81-42-984-0443, Fax: +81-42-984-0443, katagiri@saitama-med.ac.jp
Copyright © 2018 Korean Endocrine Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
- Fibrodysplasia ossificans progressiva (FOP) is a rare genetic disease that is characterized by the formation of heterotopic bone tissues in soft tissues, such as skeletal muscle, ligament, and tendon. It is difficult to remove such heterotopic bones via internal medicine or invasive procedures. The identification of activin A receptor, type I (ACVR1)/ALK2 gene mutations associated with FOP has allowed the genetic diagnosis of FOP. The ACVR1/ALK2 gene encodes the ALK2 protein, which is a transmembrane kinase receptor in the transforming growth factor-β family. The relevant mutations activate intracellular signaling in vitro and induce heterotopic bone formation in vivo. Activin A is a potential ligand that activates mutant ALK2 but not wild-type ALK2. Various types of small chemical and biological inhibitors of ALK2 signaling have been developed to establish treatments for FOP. Some of these are in clinical trials in patients with FOP.
- Fibrodysplasia ossificans progressiva (FOP) (OMIM #135100) is a rare genetic disease that is characterized by the formation of heterotopic hard tissues in soft tissues, such as skeletal muscle, ligaments, and tendons [123]. FOP is an autosomal dominant disorder with an incidence of approximately one patient per 2 million people worldwide regardless of race, location, or gender [12]. The hard tissue formed in affected patients is not simply mineralized calcium phosphate but represents the formation of new bone tissue by osteoblasts through a cartilaginous template (endochondral ossification) via the same process that is observed in normal skeletal tissues during embryonic development and regeneration. Moreover, the heterotopic bone tissue contains bone marrow and is metabolized by osteoclasts and osteoblasts in a manner similar to that observed in normal bone tissues. Heterotopic bones are distinguishable from normal bone tissues by their locations but not their biochemical characteristics, and it is therefore hard to remove only the heterotopic bone tissues in internal medicine procedure performed in patients with FOP. Although most patients with FOP can move their joints normally at birth, they show disability in various joints, including the jaw, once they reach their 30's because the heterotopic bones that formed in skeletal muscle, tendons, and ligaments gradually fuse with each other and bridge with normal bones, fixing the joints [4]. Moreover, injury to soft tissues causes acute heterotopic bone formation in patients with FOP. Thus, invasive procedures, such as biopsy, surgical operation, and injection, are prohibited in patients with FOP [5].
- Achieving a diagnosis of FOP before the onset of heterotopic ossification was difficult for a long time because there were no reliable biomarkers for this disease that could be evaluated in peripheral blood or urine. Moreover, the molecular mechanisms underlying the pathogenesis observed in patients with FOP were unknown because tissue samples were not accessible due to the contraindication for invasive procedures. The identification of the activin A receptor, type I (ACVR1)/ALK2 gene in 2006 as a gene that is responsible for FOP was a breakthrough for this rare disease [6]. The ACVR1/ALK2 gene encodes a transmembrane kinase receptor, ALK2, that binds bone morphogenetic proteins (BMPs). BMP was originally found in 1965 and described as a unique molecule in the bone matrix that induces heterotopic bone to develop in skeletal muscle [7]. The identification of a recurrent heterozygous mutation in the ACVR1/ALK2 gene in sporadic and inherited cases of FOP directly connected the BMP and FOP research fields. Moreover, those findings allowed us to examine the molecular mechanism underlying heterotopic ossification both in vitro and in vivo, to improve the diagnosis of the disease and search for potential therapeutic molecules for FOP in cultured cells and experimental animals. Because ALK2 is associated with FOP as a gain-of-function mutant receptor, various types of inhibitors have been developed to establish therapeutic drugs for FOP. Some of these molecules are in phase 2 or 3 clinical trials of patients with FOP. In this article, we review the basics of potential therapeutics for the rare disease FOP.
INTRODUCTION
- The most typical clinical feature of FOP is progressive heterotopic bone formation in soft tissues that causes disability in the joints. The progression of disability in FOP patients follows some patterns: from the upper to lower, proximal to distal and dorsal to ventral side (Table 1) [8]. The neck, spine and shoulder are the most frequently affected sites (more than 80% of cases observed until patients younger than 15 years old) [5]. In contrast, the wrists and ankles are affected in approximately 50% of patients over 30 years old. The elbows, knees, hips, and jaw become gradually affected with age until the patient reaches 40 years old. Heterotopic ossification starts in childhood and continues through adulthood but is not observed at birth. However, lumbar puncture and surgical intervention induced acute heterotopic bone formation in a 10-week-old boy [9], suggesting that co-operation between a genetic mutation and an inflammatory environment induces heterotopic bone formation in patients with FOP.
- One of the clinical features detectable at birth, before the onset of heterotopic ossification, is malformations in the big toes (Table 1). More than 90% of patients who have FOP show such malformations [51011]. An additional typical feature observed in FOP patients is tumor-like swellings [11]. Flare-ups are frequently observed before the formation of heterotopic bones is observed in soft tissues [12]. Therefore, if a patient shows both of these clinical features (malformation of the big toes and swelling), FOP should be considered before invasive procedures are performed. However, some patients with FOP do not show any clinical features before the onset of heterotopic ossification.
CLINICAL FEATURES OF FOP
- More than 90% of patients with typical FOP have an identical genetic mutation consisting of a guanine to adenine change at position 617 (c.617G>A) in the ACVR1/ALK2 gene. This causes a substitution mutation in the ALK2 protein: Arg to His at position 206 (p.R206H) (Fig. 1). Additional mutations that occur at different positions in the ACVR1/ALK2 gene have also been identified in patients with FOP with different clinical features (Fig. 1). Although some other genes were suggested to be related to FOP before the identification of the ACVR1/ALK2 gene in 2006 [6131415], no case of FOP has been shown to carry a mutation in a gene other than ACVR1/ALK2, suggesting that FOP is a typical monogenic disorder. The ACVR1/ALK2 gene is located on chromosome 2 in humans and consists of 9 coding exons. It encodes the ALK2 protein, which is a transmembrane serine/threonine (Ser/Thr) kinase receptor for members of the transforming growth factor-β (TGF-β) family (Fig. 1). Today, FOP is diagnosed by analyzing genetic mutations in the ACVR1/ALK2 gene by Sanger sequencing of polymerase chain reaction products obtained by amplifying each coding exon. Interestingly, all of the mutations identified in patients with FOP have been localized in exons 4 through 7, which encode the intracellular functional domains, the glycine/serine-rich (GS) and Ser/Thr kinase domains, both of which are important for intracellular signaling in response to ligand binding at the extracellular domain (Figs. 1, 2).
DIAGNOSIS OF FOP
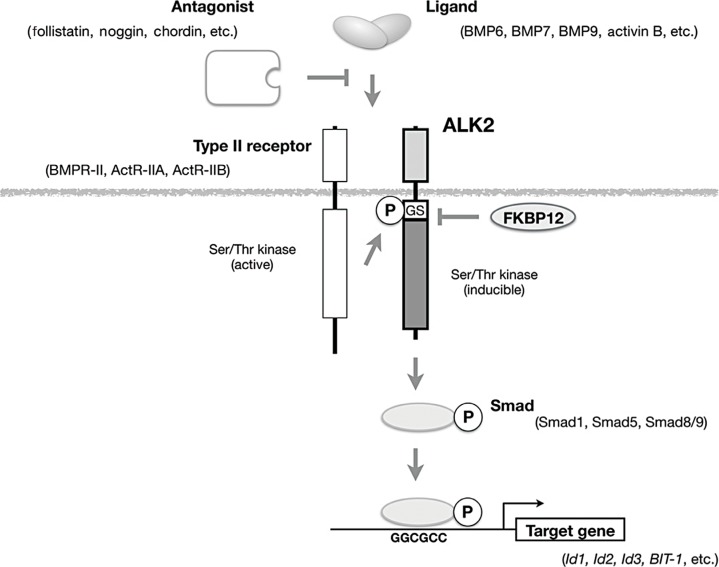
- The extracellular domain of ALK2 (a type I receptor) binds to several ligands in the TGF-β family, such as BMP-6, BMP-7, BMP9, and activin B, in co-operation with type II receptors, such as BMP receptor type II (BMPR-II), activin receptor type IIA (ActR-IIA), and activin receptor type IIB (ActR-IIB) (Fig. 2). Because type II receptors are constitutively active Ser/Thr kinases, ALK2 is phosphorylated in a ternary complex formed in response to ligand binding at the cell membrane (Fig. 2). The GS domain, which is a stretch consisting of glycine and serine residues, has been identified as the site of phosphorylation by type II receptors [16]. Phosphorylated ALK2 activates kinase activity and phosphorylates Ser and Thr residues in downstream substrates, such as Smad1, Smad5, and Smad8/9 [171819]. Phosphorylated Smad proteins regulate the transcription of target genes in the nucleus [2021].
- Transient over-expression of the mutant ALK2 associated with FOP, but not of wild-type ALK2, activates intracellular signaling without adding exogenous ligands, suggesting that these are gain-of-function mutations [22232425]. The mutant ALK2 associated with FOP is hypersensitive to the kinase activity of the type II receptors [25]. The 12 kDa FK506-binding protein (FKBP12) acts as a repressor of the kinase activity of type I receptors in the TGF-β family, including ALK2, by binding to their unphosphorylated intracellular domains [26]. It has been proposed that mutations in the intracellular domains of ALK2 associated with FOP reduce binding affinity to FKBP12 and activate downstream intracellular signaling in patients. Co-expression of the mutant ALK2 and FKBP12 in vitro reduced intracellular signaling by ALK2, but one of the mutant ALK2 associated with FOP, delP197_F198insL, was resistant to suppression by FKBP12 because it has lost the site at which it interacts with FKBP12 [27]. The clinical features of a patient carrying the mutation delP197_F198insL seemed to be similar to and not more severe than those observed in patients with typical FOP [2829]. All mutations in ALK2, including the delP197_F198insL mutation, are activated by co-expression with type II receptors, suggesting that the heterotopic ossification observed in patients is induced by intracellular signaling through mutant ALK2 in co-operation with type II receptors [2527].
- A knock-in mouse model of the p.R206H mutation showed that the phenotypes were similar to those of patients with FOP, such as malformations of the fingers and heterotopic ossification. However, they were lethal after birth [30]. A conditional knock-in mouse model with the same mutation also showed heterotopic ossification after the induction of mutant ALK2 expression [31]. In the conditional knock-in mice, heterotopic ossification was blocked by trea tment with dominant negative type II receptors, suggesting that heterotopic ossification is a ligand-dependent event [31]. Activin A is a potential ligand for heterotopic bone formation in patients with FOP because the ALK2 mutant p.R206H but not wild-type ALK2 activated intracellular signaling in response to activin A, which is not TGF-β family osteogenic ligand [3132].
- Sources of the chondrocytes and osteoblasts in heterotopic ossification were studied in vivo via a lineage tracing technique in several transgenic mouse models. Both chondrocytes and osteoblasts were differentiated from progenitor cells positive for Tie-2, a typical marker of endothelial cells [33]. Indeed, human endothelial cells underwent endothelial-mesenchyme transition following the over-expression of the p.R206H mutant of ALK2 or treatment with BMP or TGF-β in vitro [34]. During BMP-induced heterotopic bone formation in skeletal muscle tissue, both chondrocytes and osteoblasts were differentiated from Sca-1-positive interstitial mesenchymal cells but not myogenic lineage or endothelial cells [35]. Similarly, fibro/adipogenic mesenchymal cells and tendon-associated cells were identified as two types of progenitors for heterotopic ossification in an FOP mouse model that carried a mutant form of ALK2 [3637].
MOLECULAR MECHANISMS OF PATHOGENESIS IN FOP
- Because ALK2 over-signaling seemed to induce heterotopic ossification in FOP, various types of small chemical and biological inhibitors of ALK2 itself or of the up/down-stream factors in its intracellular signaling pathway have been explored (Table 2). A human anti-activin A-neutralizing antibody (REGN2477) was shown to inhibit heterotopic ossification in a mouse model with a conditional knock-in of p.R206H [31]. Using this antibody, the expansion of heterotopic ossification was also shown to be an activin A-dependent event [38]. Currently, the human anti-activin A antibody (REGN2477) is in a phase 2 clinical trial.
- Many small chemical compounds target the Ser/Thr kinase activity of ALK2. Dorsomorphin is the original compound found to act as a kinase inhibitor of type I receptors in the BMP subfamily [39]. LDN193189 was the earliest compound shown to inhibit heterotopic ossification in vivo in a mouse model of FOP [40]. Several additional kinase inhibitors have been developed to increase specificity for ALK2 among type I receptors [41424344]. However, no such compounds are currently in clinical trials.
- Some nucleic acid-based therapies have been examined in FOP. RNA interference techniques were developed to inhibit mutant but not wild-type ALK2 [4546]. An additional single nucleotide mismatch to the FOP-causing mutation increased the specificity of the mutant alleles in both the p.R206H and p.G356D mutations [45]. Exon-skipping oligo DNA was also reported to remove mutant ALK2 proteins [47].
- Chondrogenesis is inhibited by all-trans retinoic acid (RA), especially via RA receptor γ (RARγ) [48]. RARγ agonists, including palovarotene, have been shown to inhibit BMP signaling by reducing steady-state levels of Smad proteins and inhibiting heterotopic ossification in mouse models of transplantation of BMP ligands or that express mutant ALK2 in vivo [4950]. Palovarotene is currently in a phase 3 clinical trial.
- Inhibitors of BMP signaling that act through mutant ALK2 have been screened in various in vitro models. Several mammalian target of rapamycin (mTOR) inhibitors, including rapamycin, were found to be inhibitors of chondrogenesis in induced pluripotent stem (iPS) cells derived from patients with FOP who carried the p.R206H mutation [3251]. Rapamycin has been approved for and is used in other diseases, and a phase 2 clinical trial for FOP has been started. Several small chemical inhibitors of in vitro mutant ALK2 p.R206H-induced osteoblastic differentiation in C2C12 myoblasts have been reported [5253545556].
DEVELOPMENT OF NOVEL TREATMENTS FOR FOP
- In patients with FOP, heterotopic ossification is caused by gain-of-function mutations in the intracellular domains of ALK2, a type I receptor for osteogenic BMPs. The mutant ALK2 still requires ligand stimulation to induce heterotopic ossification. Various types of inhibitors of ALK2 have been studied with the aim of developing potentially effective treatments for heterotopic ossification in FOP. Some small chemicals and biological inhibitors are currently in clinical trials in patients with FOP.
CONCLUSIONS
-
Acknowledgements
- We would like to thank the members of the Division of Pathophysiology, Research Center for Genomic Medicine, Saitama Medical University and the Project of Clinical and Basic Research for FOP, Saitama Medical University for valuable discussions. Takenobu Katagiri would also like to thank Dr. Sang Wan Kim of Seoul National University College of Medicine, Korea for generously inviting us to write this review article. This work was supported in part by grants-in aid from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan (17H04317 to TK and 17K11026 to ST), a grant-in-aid from the Japan Agency for Medical Research and Development (AMED) under grant number JP17pc0101007 to Takenobu Katagiri, and a Maruki Memorial Award of Saitama Medical University to Takenobu Katagiri (No. 17-A-1-01).
ACKNOWLEDGMENTS
-
CONFLICTS OF INTEREST: Takenobu Katagiri received research grants from Daiichi-Sankyo, Co. Ltd. Sho Tsukamoto, Yutaka Nakachi, and Mai Kuratani do not have potential conflicts of interest relevant to this article.
Article information
- 1. Kaplan FS, Chakkalakal SA, Shore EM. Fibrodysplasia ossificans progressiva: mechanisms and models of skeletal metamorphosis. Dis Model Mech 2012;5:756–762. ArticlePubMedPMC
- 2. Katagiri T. Heterotopic bone formation induced by bone morphogenetic protein signaling: fibrodysplasia ossificans progressiva. J Oral Biosci 2010;52:33–41.Article
- 3. Katagiri T. A door opens for fibrodysplasia ossificans progressiva. Trends Biochem Sci 2016;41:119–121. ArticlePubMed
- 4. Shafritz AB, Shore EM, Gannon FH, Zasloff MA, Taub R, Muenke M, et al. Overexpression of an osteogenic morphogen in fibrodysplasia ossificans progressiva. N Engl J Med 1996;335:555–561. ArticlePubMed
- 5. Kitterman JA, Kantanie S, Rocke DM, Kaplan FS. Iatrogenic harm caused by diagnostic errors in fibrodysplasia ossificans progressiva. Pediatrics 2005;116:e654–e661. ArticlePubMed
- 6. Shore EM, Xu M, Feldman GJ, Fenstermacher DA, Cho TJ, Choi IH, et al. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat Genet 2006;38:525–527. ArticlePubMedPDF
- 7. Urist MR. Bone: formation by autoinduction. Science 1965;150:893–899. ArticlePubMed
- 8. Cohen RB, Hahn GV, Tabas JA, Peeper J, Levitz CL, Sando A, et al. The natural history of heterotopic ossification in patients who have fibrodysplasia ossificans progressiva. A study of forty-four patients. J Bone Joint Surg Am 1993;75:215–219. ArticlePubMed
- 9. Zaghloul KA, Heuer GG, Guttenberg MD, Shore EM, Kaplan FS, Storm PB. Lumbar puncture and surgical intervention in a child with undiagnosed fibrodysplasia ossificans progressiva. J Neurosurg Pediatr 2008;1:91–94. ArticlePubMed
- 10. Nakashima Y, Haga N, Kitoh H, Kamizono J, Tozawa K, Katagiri T, et al. Deformity of the great toe in fibrodysplasia ossificans progressiva. J Orthop Sci 2010;15:804–809. ArticlePubMed
- 11. Kaplan FS, Xu M, Glaser DL, Collins F, Connor M, Kitterman J, et al. Early diagnosis of fibrodysplasia ossificans progressiva. Pediatrics 2008;121:e1295–e1300. ArticlePubMedPMC
- 12. Pignolo RJ, Bedford-Gay C, Liljesthrom M, Durbin-Johnson BP, Shore EM, Rocke DM, et al. The natural history of flare-ups in fibrodysplasia ossificans progressiva (FOP): a comprehensive global assessment. J Bone Miner Res 2016;31:650–656. ArticlePubMed
- 13. Feldman G, Li M, Martin S, Urbanek M, Urtizberea JA, Fardeau M, et al. Fibrodysplasia ossificans progressiva, a heritable disorder of severe heterotopic ossification, maps to human chromosome 4q27-31. Am J Hum Genet 2000;66:128–135. ArticlePubMed
- 14. Lucotte G, Bathelier C, Mercier G, Gerard N, Lenoir G, Semonin O, et al. Fibrodysplasia Ossificans Progressiva Consortium. Localization of the gene for fibrodysplasia ossificans progressiva (FOP) to chromosome 17q21-22. Genet Couns 2000;11:329–334. PubMed
- 15. Xu MQ, Feldman G, Le Merrer M, Shugart YY, Glaser DL, Urtizberea JA, et al. Linkage exclusion and mutational analysis of the noggin gene in patients with fibrodysplasia ossificans progressiva (FOP). Clin Genet 2000;58:291–298. ArticlePubMed
- 16. Wrana JL, Attisano L, Wieser R, Ventura F, Massague J. Mechanism of activation of the TGF-beta receptor. Nature 1994;370:341–347. ArticlePubMedPDF
- 17. Attisano L, Wrana JL. Signal transduction by the TGF-beta superfamily. Science 2002;296:1646–1647. ArticlePubMed
- 18. Nojima J, Kanomata K, Takada Y, Fukuda T, Kokabu S, Ohte S, et al. Dual roles of Smad proteins in the conversion from myoblasts to osteoblastic cells by bone morphogenetic proteins. J Biol Chem 2010;285:15577–15586. ArticlePubMedPMC
- 19. Tsukamoto S, Mizuta T, Fujimoto M, Ohte S, Osawa K, Miyamoto A, et al. Smad9 is a new type of transcriptional regulator in bone morphogenetic protein signaling. Sci Rep 2014;4:7596ArticlePubMedPMCPDF
- 20. Katagiri T, Imada M, Yanai T, Suda T, Takahashi N, Kamijo R. Identification of a BMP-responsive element in Id1, the gene for inhibition of myogenesis. Genes Cells 2002;7:949–960. ArticlePubMed
- 21. Shin M, Ohte S, Fukuda T, Sasanuma H, Yoneyama K, Kokabu S, et al. Identification of a novel bone morphogenetic protein (BMP)-inducible transcript, BMP-inducible transcript-1, by utilizing the conserved BMP-responsive elements in the Id genes. J Bone Miner Metab 2013;31:34–43. ArticlePubMedPDF
- 22. Fukuda T, Kanomata K, Nojima J, Kokabu S, Akita M, Ikebuchi K, et al. A unique mutation of ALK2, G356D, found in a patient with fibrodysplasia ossificans progressiva is a moderately activated BMP type I receptor. Biochem Biophys Res Commun 2008;377:905–909. ArticlePubMed
- 23. Fukuda T, Kohda M, Kanomata K, Nojima J, Nakamura A, Kamizono J, et al. Constitutively activated ALK2 and increased SMAD1/5 cooperatively induce bone morphogenetic protein signaling in fibrodysplasia ossificans progressiva. J Biol Chem 2009;284:7149–7156. ArticlePubMedPMC
- 24. Shen Q, Little SC, Xu M, Haupt J, Ast C, Katagiri T, et al. The fibrodysplasia ossificans progressiva R206H ACVR1 mutation activates BMP-independent chondrogenesis and zebrafish embryo ventralization. J Clin Invest 2009;119:3462–3472. ArticlePubMedPMC
- 25. Fujimoto M, Ohte S, Osawa K, Miyamoto A, Tsukamoto S, Mizuta T, et al. Mutant activin-like kinase 2 in fibrodysplasia ossificans progressiva are activated via T203 by BMP type II receptors. Mol Endocrinol 2015;29:140–152. ArticlePubMedPDF
- 26. Wang T, Li BY, Danielson PD, Shah PC, Rockwell S, Lechleider RJ, et al. The immunophilin FKBP12 functions as a common inhibitor of the TGF beta family type I receptors. Cell 1996;86:435–444. ArticlePubMed
- 27. Machiya A, Tsukamoto S, Ohte S, Kuratani M, Fujimoto M, Kumagai K, et al. Effects of FKBP12 and type II BMP receptors on signal transduction by ALK2 activating mutations associated with genetic disorders. Bone 2018;111:101–108. ArticlePubMed
- 28. Kaplan FS, Xu M, Seemann P, Connor JM, Glaser DL, Carroll L, et al. Classic and atypical fibrodysplasia ossificans progressiva (FOP) phenotypes are caused by mutations in the bone morphogenetic protein (BMP) type I receptor ACVR1. Hum Mutat 2009;30:379–390. ArticlePubMedPMC
- 29. Gregson CL, Hollingworth P, Williams M, Petrie KA, Bullock AN, Brown MA, et al. A novel ACVR1 mutation in the glycine/serine-rich domain found in the most benign case of a fibrodysplasia ossificans progressiva variant reported to date. Bone 2011;48:654–658. ArticlePubMed
- 30. Chakkalakal SA, Zhang D, Culbert AL, Convente MR, Caron RJ, Wright AC, et al. An Acvr1 R206H knock-in mouse has fibrodysplasia ossificans progressiva. J Bone Miner Res 2012;27:1746–1756. ArticlePubMedPMC
- 31. Hatsell SJ, Idone V, Wolken DM, Huang L, Kim HJ, Wang L, et al. ACVR1R206H receptor mutation causes fibrodysplasia ossificans progressiva by imparting responsiveness to activin A. Sci Transl Med 2015;7:303ra137.ArticlePubMedPMC
- 32. Hino K, Ikeya M, Horigome K, Matsumoto Y, Ebise H, Nishio M, et al. Neofunction of ACVR1 in fibrodysplasia ossificans progressiva. Proc Natl Acad Sci U S A 2015;112:15438–15443. ArticlePubMedPMC
- 33. Lounev VY, Ramachandran R, Wosczyna MN, Yamamoto M, Maidment AD, Shore EM, et al. Identification of progenitor cells that contribute to heterotopic skeletogenesis. J Bone Joint Surg Am 2009;91:652–663. ArticlePubMedPMC
- 34. Medici D, Shore EM, Lounev VY, Kaplan FS, Kalluri R, Olsen BR. Conversion of vascular endothelial cells into multipotent stem-like cells. Nat Med 2010;16:1400–1406. ArticlePubMedPMCPDF
- 35. Wosczyna MN, Biswas AA, Cogswell CA, Goldhamer DJ. Multipotent progenitors resident in the skeletal muscle interstitium exhibit robust BMP-dependent osteogenic activity and mediate heterotopic ossification. J Bone Miner Res 2012;27:1004–1017. ArticlePubMedPMC
- 36. Dey D, Bagarova J, Hatsell SJ, Armstrong KA, Huang L, Ermann J, et al. Two tissue-resident progenitor lineages drive distinct phenotypes of heterotopic ossification. Sci Transl Med 2016;8:366ra163.ArticlePubMedPMC
- 37. Lees-Shepard JB, Yamamoto M, Biswas AA, Stoessel SJ, Nicholas SE, Cogswell CA, et al. Activin-dependent signaling in fibro/adipogenic progenitors causes fibrodysplasia ossificans progressiva. Nat Commun 2018;9:471ArticlePubMedPMC
- 38. Upadhyay J, Xie L, Huang L, Das N, Stewart RC, Lyon MC, et al. The expansion of heterotopic bone in fibrodysplasia ossificans progressiva is activin a-dependent. J Bone Miner Res 2017;32:2489–2499. ArticlePubMed
- 39. Yu PB, Hong CC, Sachidanandan C, Babitt JL, Deng DY, Hoyng SA, et al. Dorsomorphin inhibits BMP signals required for embryogenesis and iron metabolism. Nat Chem Biol 2008;4:33–41. ArticlePubMedPDF
- 40. Yu PB, Deng DY, Lai CS, Hong CC, Cuny GD, Bouxsein ML, et al. BMP type I receptor inhibition reduces heterotopic [corrected] ossification. Nat Med 2008;14:1363–1369. ArticlePubMedPMCPDF
- 41. Hao J, Ho JN, Lewis JA, Karim KA, Daniels RN, Gentry PR, et al. In vivo structure-activity relationship study of dorsomorphin analogues identifies selective VEGF and BMP inhibitors. ACS Chem Biol 2010;5:245–253. ArticlePubMedPMC
- 42. Mohedas AH, Xing X, Armstrong KA, Bullock AN, Cuny GD, Yu PB. Development of an ALK2-biased BMP type I receptor kinase inhibitor. ACS Chem Biol 2013;8:1291–1302. ArticlePubMedPMC
- 43. Sanvitale CE, Kerr G, Chaikuad A, Ramel MC, Mohedas AH, Reichert S, et al. A new class of small molecule inhibitor of BMP signaling. PLoS One 2013;8:e62721. ArticlePubMedPMC
- 44. Engers DW, Frist AY, Lindsley CW, Hong CC, Hopkins CR. Synthesis and structure-activity relationships of a novel and selective bone morphogenetic protein receptor (BMP) inhibitor derived from the pyrazolo[1.5-a]pyrimidine scaffold of dorsomorphin: the discovery of ML347 as an ALK2 versus ALK3 selective MLPCN probe. Bioorg Med Chem Lett 2013;23:3248–3252. ArticlePubMedPMC
- 45. Takahashi M, Katagiri T, Furuya H, Hohjoh H. Disease-causing allele-specific silencing against the ALK2 mutants, R206H and G356D, in fibrodysplasia ossificans progressiva. Gene Ther 2012;19:781–785. ArticlePubMedPDF
- 46. Kaplan J, Kaplan FS, Shore EM. Restoration of normal BMP signaling levels and osteogenic differentiation in FOP mesenchymal progenitor cells by mutant allele-specific targeting. Gene Ther 2012;19:786–790. ArticlePubMedPDF
- 47. Shi S, Cai J, de Gorter DJ, Sanchez-Duffhues G, Kemaladewi DU, Hoogaars WM, et al. Antisense-oligonucleotide mediated exon skipping in activin-receptor-like kinase 2: inhibiting the receptor that is overactive in fibrodysplasia ossificans progressiva. PLoS One 2013;8:e69096. ArticlePubMedPMC
- 48. Shimono K, Morrison TN, Tung WE, Chandraratna RA, Williams JA, Iwamoto M, et al. Inhibition of ectopic bone formation by a selective retinoic acid receptor alpha-agonist: a new therapy for heterotopic ossification? J Orthop Res 2010;28:271–277. ArticlePubMed
- 49. Shimono K, Tung WE, Macolino C, Chi AH, Didizian JH, Mundy C, et al. Potent inhibition of heterotopic ossification by nuclear retinoic acid receptor-γ agonists. Nat Med 2011;17:454–460. ArticlePubMedPMCPDF
- 50. Chakkalakal SA, Uchibe K, Convente MR, Zhang D, Economides AN, Kaplan FS, et al. Palovarotene inhibits heterotopic ossification and maintains limb mobility and growth in mice with the human ACVR1(R206H) fibrodysplasia ossificans progressiva (FOP) mutation. J Bone Miner Res 2016;31:1666–1675. ArticlePubMedPMC
- 51. Hino K, Horigome K, Nishio M, Komura S, Nagata S, Zhao C, et al. Activin-A enhances mTOR signaling to promote aberrant chondrogenesis in fibrodysplasia ossificans progressiva. J Clin Invest 2017;127:3339–3352. ArticlePubMedPMC
- 52. Fukuda T, Uchida R, Inoue H, Ohte S, Yamazaki H, Matsuda D, et al. Fungal pyrrolidine-containing metabolites inhibit alkaline phosphatase activity in bone morphogenetic protein-stimulated myoblastoma cells. Acta Pharm Sin B 2012;2:23–27.Article
- 53. Fukuda T, Uchida R, Ohte S, Inoue H, Yamazaki H, Matsuda D, et al. Trichocyalides A and B, new inhibitors of alkaline phosphatase activity in bone morphogenetic protein-stimulated myoblasts, produced by Trichoderma sp. FKI-5513. J Antibiot (Tokyo) 2012;65:565–569. ArticlePubMedPDF
- 54. Yamamoto R, Matsushita M, Kitoh H, Masuda A, Ito M, Katagiri T, et al. Clinically applicable antianginal agents suppress osteoblastic transformation of myogenic cells and heterotopic ossifications in mice. J Bone Miner Metab 2013;31:26–33. ArticlePubMedPDF
- 55. Uchida R, Nakai M, Ohte S, Onaka H, Katagiri T, Tomoda H. 5-Prenyltryptophol, a new inhibitor of bone morphogenetic protein-induced alkaline phosphatase expression in myoblasts, produced by Streptomyces colinus subsp. Albescens HEK608. J Antibiot (Tokyo) 2014;67:589–591. ArticlePubMedPDF
- 56. Uchida R, Lee D, Suwa I, Ohtawa M, Watanabe N, Demachi A, et al. Scopranones with two atypical scooplike moieties produced by streptomyces sp. BYK-11038. Org Lett 2017;19:5980–5983. ArticlePubMed
References
Schematic representation of the relationship between the activin A receptor, type I (ACVR1)/ALK2 gene, complementary DNA (cDNA) and protein. The ACVR1/ALK2 gene consist of 9 coding exons (Ex.) (black boxes). The ACVR1/ALK2 cDNA (1,530 bp) encodes a protein with 509 amino acids (a. a.). Mutations associated with fibrodysplasia ossificans progressiva are shown in the figure. The positions of the mutations in the cDNA and protein are indicated by numbers that begin from the adenine of the first ATG codon and Met residue, respectively. TGA, stop codon; SP, signal peptide; TM, transmembrane domain; GS, glycine/serine-rich domain; Ser/Thr kinase, serine/threonine kinase domain.

Schematic representation of signal transduction by ALK2 in response to ligand binding. ALK2 binds to a transforming growth factor-β family ligand, such as bone morphogenetic protein 6 (BMP6), BMP7, and BMP9, and acts as a type I receptor in co-operation with one of the type II receptors (BMP receptor type II [BMPR-II], activin receptor type IIA [ActR-IIA], and activin receptor type IIB [ActR-IIB]). Antagonists, such as follistatin, noggin, and chordin, directly bind to the ligand and prevent it from binding to receptors. Type II receptors are constitutively active kinases that phosphorylate the glycine/serine-rich domain (GS) domain of ALK2 to activate kinase activity. Activated ALK2 phosphorylates downstream substrates, such as Smad1, Smad5, and Smad8/9, and then binds to specific DNA sequences to regulate the transcription of its target genes. Ser/Thr, serine/threonine; P, phosphorylation; FKBP12, 12 kDa FK506-binding protein; Id1, inhibitor of DNA binding 1; BIT-1, BMP-inducible transcript-1.

Major Features of Patients with Fibrodysplasia Ossificans Progressiva
Inhibitors of ALK2 Signaling
| Target | Molecule | Clinical trial | Reference |
|---|---|---|---|
| Ligand | Anti-activin A antibody (REGN2477) | Phase 2 | [31] |
| sActR-IIA-Fc | [31] | ||
| sActR-IIB-Fc | [31] | ||
| ALK2 kinase | Dorsomorphin | [39] | |
| LDN193189 | [40] | ||
| LDN212854 | [42] | ||
| K02288 | [43] | ||
| DMH-1 | [41] | ||
| ML347 | [44] | ||
| ALK2 mRNA | Allele specific RNAi | [4546] | |
| Exon skipping oligo DNA | [47] | ||
| Smads | RARγ agonist (palovarotene) | Phase 3 | [4950] |
| All-trans retinoic acid | [48] | ||
| BMP signaling | Fendiline | [54] | |
| Perhexiline | [54] | ||
| Differentiation | Rapamycin | Phase 2 | [51] |
| NG-391 | [52] | ||
| NG-393 | [52] | ||
| Trichocyalide A | [53] | ||
| Trichocyalide B | [53] |
Figure & Data
References
Citations
- How Activin A Became a Therapeutic Target in Fibrodysplasia Ossificans Progressiva
Dushyanth Srinivasan, Martin Arostegui, Erich J. Goebel, Kaitlin N. Hart, Senem Aykul, John B. Lees-Shepard, Vincent Idone, Sarah J. Hatsell, Aris N. Economides
Biomolecules.2024; 14(1): 101. CrossRef - Fibrodysplasia Ossificans Progressiva Mimics Generalized Dystonia Disorder: A Case Report
Seraj Makkawi, Osama Khojah, Reema Abualnaja, Abdulaziz Qashqari, Nawaf A Alahmadi, Abdullatif G Bshnaq, Abdulrahman Alharthi, Hashem H Al-Hashemi, Aiman M Shawli
Cureus.2023;[Epub] CrossRef - Exploration of marine natural resources in Indonesia and development of efficient strategies for the production of microbial halogenated metabolites
Hiroyuki Yamazaki
Journal of Natural Medicines.2022; 76(1): 1. CrossRef - A Novel De Novo Frameshift Pathogenic Variant in the FAM111B Resulting in Progressive Osseous Heteroplasia Phenotype
Anna Ryabets-Lienhard, Panadeekarn Panjawatanan, Kyle Vogt, Jianling Ji, Senta Georgia, Pisit Pitukcheewanont
Calcified Tissue International.2022; 112(4): 518. CrossRef - Inhibitory effects of sesquiterpene lactones from the Indonesian marine sponge Lamellodysidea cf. herbacea on bone morphogenetic protein-induced osteoblastic differentiation
Satoshi Ohte, Hiroyuki Yamazaki, Ohgi Takahashi, Henki Rotinsulu, Defny S. Wewengkang, Deiske A. Sumilat, Delfly B. Abdjul, Wilmar Maarisit, Magie M. Kapojos, Huiping Zhang, Fumiaki Hayashi, Michio Namikoshi, Takenobu Katagiri, Hiroshi Tomoda, Ryuji Uchid
Bioorganic & Medicinal Chemistry Letters.2021; 35: 127783. CrossRef - Genomic Context and Mechanisms of the ACVR1 Mutation in Fibrodysplasia Ossificans Progressiva
Roberto Ravazzolo, Renata Bocciardi
Biomedicines.2021; 9(2): 154. CrossRef - New insights on fibrodysplasia ossificans progressiva: discussion of an autoptic case report and brief literature review
Vittorio Bolcato, Claudia Carelli, Silvia Damiana Visonà, Marcella Reguzzoni, Maja Di Rocco, Alessandra Radogna, Livio Pietro Tronconi, Matteo Moretti
Intractable & Rare Diseases Research.2021; 10(2): 136. CrossRef - Accumulated Knowledge of Activin Receptor-Like Kinase 2 (ALK2)/Activin A Receptor, Type 1 (ACVR1) as a Target for Human Disorders
Takenobu Katagiri, Sho Tsukamoto, Mai Kuratani
Biomedicines.2021; 9(7): 736. CrossRef - Cytoskeleton Reorganization in EndMT—The Role in Cancer and Fibrotic Diseases
Wojciech Michał Ciszewski, Marta Ewelina Wawro, Izabela Sacewicz-Hofman, Katarzyna Sobierajska
International Journal of Molecular Sciences.2021; 22(21): 11607. CrossRef - Alendronate disturbs femoral growth due to changes during immunolocalization of transforming growth factor-β1 and bone morphogenetic protein-2 in epiphyseal plate
Juliana Souza Vieira, Emanuelle Juliana Cunha, Juliana Feltrin de Souza, Luis Henrique Koeler Chaves, Jessica Lakes de Souza, Allan Fernando Giovanini
World Journal of Experimental Medicine.2020; 10(1): 1. CrossRef - ALK2: A Therapeutic Target for Fibrodysplasia Ossificans Progressiva and Diffuse Intrinsic Pontine Glioma
Katsuhiko Sekimata, Tomohiro Sato, Naoki Sakai
Chemical and Pharmaceutical Bulletin.2020; 68(3): 194. CrossRef - Role of Signal Transduction Pathways and Transcription Factors in Cartilage and Joint Diseases
Riko Nishimura, Kenji Hata, Yoshifumi Takahata, Tomohiko Murakami, Eriko Nakamura, Maki Ohkawa, Lerdluck Ruengsinpinya
International Journal of Molecular Sciences.2020; 21(4): 1340. CrossRef - Design of primers for direct sequencing of nine coding exons in the human ACVR1 gene
Masaru Matsuoka, Sho Tsukamoto, Yuta Orihara, Rieko Kawamura, Mai Kuratani, Nobuhiko Haga, Kenji Ikebuchi, Takenobu Katagiri
Bone.2020; 138: 115469. CrossRef - A new diketopiperazine-like inhibitor of bone morphogenetic protein-induced osteoblastic differentiation produced by marine-derived Aspergillus sp. BFM-0085
Satoshi Ohte, Takehiro Shiokawa, Nobuhiro Koyama, Takenobu Katagiri, Chiaki Imada, Hiroshi Tomoda
The Journal of Antibiotics.2020; 73(8): 554. CrossRef - Penicillic Acid Congener, a New Inhibitor of BMP-Induced Alkaline Phosphatase Activity in Myoblasts, Produced by the Fungus Penicillium sp. BF-0343
Nobuhiro Koyama, Yasuhiro Otoguro, Satoshi Ohte, Takenobu Katagiri, Hiroshi Tomoda
Natural Product Communications.2020; 15(9): 1934578X2094265. CrossRef - Fibrodysplasia ossificans progressiva: current concepts from bench to bedside
Arun-Kumar Kaliya-Perumal, Tom J. Carney, Philip W. Ingham
Disease Models & Mechanisms.2020;[Epub] CrossRef - Clinical Aspects and Current Therapeutic Approaches for FOP
Hiroshi Kitoh
Biomedicines.2020; 8(9): 325. CrossRef - Screening for Small Molecule Inhibitors of BMP-Induced Osteoblastic Differentiation from Indonesian Marine Invertebrates
Hiroyuki Yamazaki, Satoshi Ohte, Henki Rotinsulu, Defny S. Wewengkang, Deiske A. Sumilat, Delfly B. Abdjul, Wilmar Maarisit, Magie M. Kapojos, Michio Namikoshi, Takenobu Katagiri, Hiroshi Tomoda, Ryuji Uchida
Marine Drugs.2020; 18(12): 606. CrossRef - Propranolol and ascorbic acid in control of fibrodysplasia ossificans progressiva flare-ups due to accidental falls
Durval Batista Palhares, Deborah Ribeiro Nascimento, Marilene Garcia Palhares, Suzana Lopes Bomfim Balaniuc, Liane de Rosso Giuliani, Paula Cristhina Niz Xavier, José Mauro Goulart Brum, Fabiana Alves, Francisco Oliveira Vieira, Elaine Maria Souza-Fagunde
Intractable & Rare Diseases Research.2019; 8(1): 24. CrossRef - Late-onset fibrodysplasia ossificans progressiva with atypical presentation: A case report
Conor M. Cunningham, J. Matthew Royeca, Samuel W. King, Hemant Pandit
Case Reports in Women's Health.2019; 23: e00134. CrossRef - Fibrodysplasia ossificans progressiva: lessons learned from a rare disease
Gulseren Akyuz, Kardelen Gencer-Atalay, Pinar Ata
Current Opinion in Pediatrics.2019; 31(6): 716. CrossRef - Discovery of Heterotopic Bone-Inducing Activity in Hard Tissues and the TGF-β Superfamily
Takenobu Katagiri, Sho Tsukamoto, Yutaka Nakachi, Mai Kuratani
International Journal of Molecular Sciences.2018; 19(11): 3586. CrossRef
 PubReader
PubReader ePub Link
ePub Link Cite
Cite