
Articles
- Page Path
- HOME > Endocrinol Metab > Volume 34(1); 2019 > Article
-
Review ArticleMouse Models as a Tool for Understanding Progression in BrafV600E-Driven Thyroid Cancers
-
Iñigo Landa1
 , Jeffrey A. Knauf1,2
, Jeffrey A. Knauf1,2 -
Endocrinology and Metabolism 2019;34(1):11-22.
DOI: https://doi.org/10.3803/EnM.2019.34.1.11
Published online: February 15, 2019
1Human Oncology and Pathogenesis Program, Memorial Sloan Kettering Cancer Center, New York, NY, USA.
2Department of Medicine, Memorial Sloan Kettering Cancer Center, New York, NY, USA.
- Corresponding author: Jeffrey A. Knauf. Human Oncology and Pathogenesis Program, Department of Medicine, Memorial Sloan Kettering Cancer Center, 1275 York Avenue, New York, NY 10065, USA. Tel: +1-1-646-888-2164, Fax: +1-1-646-422-0675, Knaufj@mskcc.org
Copyright © 2019 Korean Endocrine Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
- The development of next generation sequencing (NGS) has led to marked advancement of our understanding of genetic events mediating the initiation and progression of thyroid cancers. The NGS studies have confirmed the previously reported high frequency of mutually-exclusive oncogenic alterations affecting BRAF and RAS proto-oncogenes in all stages of thyroid cancer. Initially identified by traditional sequencing approaches, the NGS studies also confirmed the acquisition of alterations that inactivate tumor protein p53 (TP53) and activate phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA) in advanced thyroid cancers. Novel alterations, such as those in telomerase reverse transcriptase (TERT) promoter and mating-type switching/sucrose non-fermenting (SWI/SNF) complex, are also likely to promote progression of the BRAFV600E-driven thyroid cancers. A number of genetically engineered mouse models (GEMM) of BRAFV600E-driven thyroid cancer have been developed to investigate thyroid tumorigenesis mediated by oncogenic BRAF and to explore the role of genetic alterations identified in the genomic analyses of advanced thyroid cancer to promote tumor progression. This review will discuss the various GEMMs that have been developed to investigate oncogenic BRAFV600E-driven thyroid cancers.
- Cancers that originate from thyroid follicular cells, which are the cells in the thyroid responsible for producing thyroid hormones, can be classified into two main groups: differentiated and undifferentiated. The differentiated consist of follicular and papillary, the latter of which account for the vast majority of thyroid cancer cases. In many cases there is a step-wise progression from differentiated thyroid cancers to undifferentiated cancers. The progression is accompanied by a marked difference in prognosis. Most differentiated cancers are well controlled through surgery and radioiodine therapy. By contrast, the undifferentiated cancers do not respond to radioiodine and commonly have metastasis, limiting the effectiveness of surgery. The gainof-function BRAFV600E is the most common oncogenic driver in thyroid cancers. The canonical signaling pathway activated by BRAF is thought to result in the near exclusive activation of MEK and ERK. Most undifferentiated thyroid cancers are thought to arise from preexisting well-differentiated tumors as a result of acquiring additional alterations. Genomic analysis of advanced thyroid cancers has identified a number of genes that cooperate with oncogenic BRAF to promote thyroid cancer progression. Using the genomics of thyroid cancer to guide the development of mouse models of thyroid cancers has led to marked improvement in our understanding of thyroid cancer progression and how the additional alterations that promote thyroid cancer progression might influence their response to therapy. Here we review the genomics of advanced thyroid cancers driven by BRAFV600E and the various genetically engineered mouse models of oncogenic BRAF-driven thyroid cancer.
INTRODUCTION
- In the recent years, several studies in large cohorts of patients, encompassing the full spectrum of human thyroid cancers, from well-differentiated to undifferentiated, have defined the genomic landscape of these tumors [12345]. These reports provided snapshots of the genetic makeup of papillary thyroid carcinoma (PTC), poorly-differentiated thyroid carcinoma (PDTC), and anaplastic thyroid cancers (ATCs), and gave insights into the key genetic alterations that promote thyroid cancer progression. Mutations in BRAFV600E are tumor-initiating events, occur in roughly half of all thyroid tumors, and are common in both well-differentiated (such as PTC) and advanced cancers (PDTC and ATC). Table 1 and Fig. 1 summarize the crucial genetic events that distinguish PTCs from PDTCs and ATCs. The role of some of these alterations in thyroid cancer progression has been demonstrated in vitro and in vivo (e.g., tumor protein p53 [TP53] loss-of-function, activation of phosphatidylinositol-4,5-bisphosphate 3-kinase [PI3K]/AKT pathway), whereas the precise mechanisms by which others contribute to this process remains to be tested.
- Mutations in TP53 gene, which typically inactivate the p53 protein, are extremely rare events in PTCs, whereas they occur in at least 50% of ATCs. Hotspot mutations in the proximal promoter of the telomerase reverse transcriptase (TERT) gene are the second most consistently associated genetic alteration in thyroid cancer progression: they occur in 11% of BRAFV600E-mutant PTCs, 44% of PDTCs, and 55% of ATCs. The fact that most of TERT mutations in PTCs are subclonal, whereas all of them are clonal events in PDTCs and ATCs suggests that these lesions are selected during thyroid cancer progression. Activating mutations in phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA), usually affecting key residues in the protein's helical (E542K, E545K) and kinase (H1047R) domains are strongly associated with BRAFV600E-mutant tumors, and track with thyroid cancer progression. The former points to activation of PI3K/AKT pathway as an important process in these tumors, further reinforced by the occasional presence of AKT1 and AKT2 oncogenic mutations (e.g., E17K). However, the observation that truncating mutations in phosphatase and tensin homolog (PTEN) gene are relatively common in non-BRAFV600E advanced thyroid tumors but mutually-exclusive with their BRAFV600E-mutant counterparts suggests that the precise mechanism by which PI3K/AKT pathway is activated matters in tumor biology.
- Mutations in members of the SWI/SNF chromatin remodeling complex are significantly enriched in ATCs versus PTCs, both in BRAFV600E-mutant and BRAF wild-type tumors. These are generally mutually-exclusive truncating mutations on genes such as AT-rich interaction domain 1A (ARID1A), ARID1B, ARID2, SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily b, member 1 (SMARCB1), and polybromo 1 (PBRM1). The exact role of aberrant SWI/SNF function in thyroid cancer biology remains to be tested. Truncating events affecting cell cycle regulator cyclin dependent kinase inhibitor 2A (CDKN2A) are also enriched in ATCs, and this is further supported by the frequent copy number losses affecting chr9p21.3 locus, which spans CDKN2A gene (not shown). Other less-frequent genetic events, such as truncating mutations targeting neurofibromin 2 (NF2) and RNA binding motif protein 10 (RBM10) genes, appear to be enriched in BRAFV600E-mutant ATCs as well.
- Finally, it is reasonable to think that the generalization of whole-exome and whole-genome sequencing technologies will eventually unmask novel alterations contributing to BRAFV600E-mutant thyroid cancer progression, but these will likely affect a small proportion of cases.
GENOMICS OF BRAFV600E-DRIVEN THYROID CANCERS
- A large number of studies (reviewed in [6]), including the next generation sequencing studies of advanced thyroid cancers described above, have demonstrated that carcinomas arising from thyroid follicular cells have non-overlapping activating mutations of the growth factor receptors RET or neurotrophic receptor tyrosine kinase 1/3 (NTRK1/3), the three isoforms of RAS (N, H, and K), or of BRAF, which altogether are found in approximately 70% of cases. The fact that all of these alterations activate the mitogen-activated protein kinase (MAPK) pathway highlights its importance in thyroid cancer biology. This has been confirmed through pharmacological and genetic targeting of BRAF and MEK in both preclinical [78910] and clinical studies [1112]. As BRAFV600E is the most common alteration that activates the MAPK pathway in thyroid cancers, a number of mouse models of thyroid cancers driven by oncogenic Braf have been developed (Table 2) [8131415161718192021222324], providing valuable insight into the biology of thyroid cancer driven by this oncoprotein. In the subsequent sections the various mouse models are discussed.
THYROID CANCERS AND THE MAPK PATHWAY
- The identification of BRAFV600E in papillary thyroid microcarcinoma (reviewed in [25]) suggests that it is likely an initiating event in thyroid cancer. This hypothesis was first tested by Knauf et al. [13], using the bovine thyroglobulin promoter to drive thyroid-specific expression of a human BRAFV600E cDNA (Tg-Braf). In this study, two mouse lines were created, Tg-BRAF2 and Tg-BRAF3. Both lines developed PTCs, albeit with different latency and penetrance, which was attributed to differences in BRAFV600E expression, confirming that oncogenic BRAF alone was able to promote PTC development [13]. Similar results were obtained by Rusinek et al. [14] who generated three different lines with thyroglobulin-driven expression of human influenza hemagglutinin (HA)-tagged human BRAFV600E. In these two models, BRAFV600E was overexpressed and not under control of its endogenous promoter. In addition, expression of the oncoprotein promoted thyroid dedifferentiation resulting in decreased expression of genes involved in thyroid hormone production, which includes Tg. Since the Tg promoter drives expression of the transgenic BRAF in these models, it is likely that expression of the oncoprotein attenuates its own expression, creating a complex negative feedback loop. To overcome this, Franco et al. [15], used lox-stop-lox (LSL)-BrafV600E mice which contains a Cre-regulated conditional BrafV600E knock-in allele. Crossing these mice with thyroid peroxidase (TPO)-Cre mice generated animals with thyroid-specific expression of BrafV600E under control of the endogenous Braf promoter, which is expected to begin around embryonic day 14. The resulting LSL-BrafV600E/TPO-Cre mice developed PTCs with a near 100% penetrance at 5 weeks, confirming oncogenic Braf even at endogenous levels is sufficient to promote thyroid cancer. This was confirmed by Charles et al. [16], who crossed mice harboring a Cre-activated BrafV600E allele (BrafCA) with Tg-CreERT2 mice to create Thyro/CreERT2/BrafCA mice with inducible, thyroid-specific knock-in of BrafV600E. Fourteen days after BrafV600E expression (upon treatment with tamoxifen), thyrocytes displayed a squamous morphology accompanied by a large increase in follicle size, and after 3 months, occasional foci of hyperplastic tall cells were detected, but it was not until 6 months that PTC was detected. The extended tumor latency in the Thyro/CreERT2/BrafCA mice compared to LSL-BrafV600E/TPO-Cre is likely due to the activation of Braf when animals are adults, whereas it occurs during embryonic development in the LSL-BrafV600E/TPO-Cre mice. However, subtle differences between the BrafCA and LSL-BrafV600E alleles cannot be ruled out.
- In all of the models above, thyroid-specific expression of oncogenic BRAF was associated with decreased thyroid function and an increase in serum thyroid-stimulating hormone (TSH) levels, raising the possibility that signaling via the TSH receptor cooperates with oncogenic Braf. Indeed, Franco et al. [15] demonstrated that crossing the LSL-BrafV600E/TPO-Cre mice with TSH receptor knockout mice resulted in a marked delay in PTC formation. However, suppression of TSH by high doses of T4 starting shortly after birth, when the thyroid still had a normal histology, was not able to suppress tumorigenesis [15]. The reason for this was not investigated but could possibly be because T4 was administered after the Braf-expressing cells had already become refractory to TSH action. Alternatively, since unliganded TSH receptor retains significant constitutive activity [26], absence of ligand alone may not be sufficient to block the pathway. Consistent with amplified TSH receptor signaling not being required for BrafV600E-induced thyroid tumorigenesis is the observation that focal activation of BrafV600E in the thyroid of BrafCA mice by either leaky Cre activity from the Tg-CreERT2 allele [16] or thyroid injection of adenovirus expressing Cre recombinase under the control of the thyroglobulin promoter [17] resulted in PTCs despite not elevating TSH.
- Expression of BRAFV600E in the immortal differentiated rat thyroid cell line, PCCL3, blocks thyroid differentiation, which can be reversed by inhibition of the MAPK pathway [2728]. This observation was first confirmed in vivo by Chakravarty et al. [18], who demonstrated that thyroid-specific, dox-inducible expression of BRAFV600E blocked thyroid differentiation and generated radioactive iodide (RAI)-refractory PTCs. Consistent with in vitro data, this was completely reversible by genetic inhibition of BRAFV600E, and partially by pharmacological inhibition of RAF or MEK [18]. These data ultimately led to a clinical trial testing the MEK inhibitor selumetinib in RAI-refractory thyroid cancers, demonstrating restoration of iodide uptake that was sufficient to justify treating with RAI in eight of 20 patients [29]. Selumetinib restored uptake in all five patients harboring a NRAS mutation, but only in one of nine BRAFV600E-positive patients. The difference observed between RAS-mutant and BRAF-mutant tumors was suggested to be due to incomplete inhibition of the MAPK pathway in the BRAF-mutant tumors as a result of higher flux through the pathway. This hypothesis was supported by Nagarajah et al. [19], who demonstrated selumetinib was ineffective at restoring thyroid differentiation in LSL-BrafV600E/TPO-Cre mice, but the more potent MEK inhibitor CH5126766 was able to restore differentiation and responsiveness to RAI. CH5126766 is an allosteric inhibitor of MEK that, upon binding, places MEK in an inactive complex with RAF, and thus functions as a RAF/MEK dual inhibitor [30]. In human clinical trials, the RAF inhibitors dabrafenib and vemurafenib, which are more effective than selumetinib in blocking the MAPK pathway, restored iodide uptake sufficiently to justify treating with RAI in six of 10 patients and four of 10 BRAFV600E-mutant patients, respectively [3132].
MOUSE MODELS OF BRAFV600E-INDUCED PTCs
- Alterations in effectors of the PI3K pathway are found in approximately 38% of advanced thyroid cancer harboring BRAFV600E (Table 1, Fig. 1). Activating mutations of PIK3CA (32%) are the most common, followed by AKT1 and AKT2 (Fig. 1) [245]. While inactivating alterations in PTEN are found in advanced thyroid cancer, they are typically associated with activating mutations of RAS or inactivation of NF1, and are very rarely found in BRAFV600E mutant thyroid cancers [245]. Charles et al. [20], crossed Tg-CreERT2/BRAFCA mice with Pik3caH1047R mice to generate mice with tamoxifen-induced, thyroid-specific knock-in of BrafV600E and Pik3caH1047R. Tamoxifen-induced expression of Pik3caH1047R alone produced no thyroid phenotype even 1 year after tamoxifen-induced activation. By contrast, expression of BrafV600E alone produced PTCs 6 to 9 months after BRAF activation. Combining the two oncoproteins hastened BrafV600E-induced PTC formation (PTC detectable 3 to 6 months after tamoxifen), accelerated tumor growth, and after 1 year 70% of the Tg-CreERT2/BrafCA/Pik3caH1047R mice required euthanasia due to labored breathing and thyroid tumors >1 cm3. Histological examination showed 80% of tumors exhibited features consistent with progression to ATCs, which was not seen in tumors expressing BrafV600E alone. Charles et al. [20], further confirmed the accelerated progression of BrafV600E-driven thyroid cancers after an alternative intervention for Pi3k pathway activation by crossing Tg-CreERT2/BrafCA with Ptenflox/flox mice which, like Pik3caH1047R, showed accelerated PTC development and progression to ATC. Shimamura et al. [17] demonstrated that activation of BrafV600E and deleting a single allele of PTEN induced PTCs with undifferentiated foci whereas BrafV600E activation alone produced PTCs without an undifferentiated component. In these experiments, the oncogenic changes were induced by injection of adenovirus expressing Cre under the control of the thyroid globulin promoter into a single thyroid lobe of BrafCA/+/Ptenflox/+ or BrafCA/+ mice. Very weak Pten staining in the resulting tumors from the BrafCA/+/Ptenflox/+ mice, which was readily detected in PTCs from BrafCA/+, suggests that expression from the second Pten allele was suppressed during tumor progression. Consistent with a more aggressive tumor, 67% of the Braf-CA/+/Ptenflox/+ mice were found to have microscopic lung metastasis versus only 22% in the BrafCA/+ mice. The mutually-exclusive occurrence of BRAF and PTEN alterations in human thyroid cancers limits the usefulness of Braf/Pten mice to test drug combinations.
- In melanomas, activation of the PI3K pathway was shown to promote resistance to RAF inhibitors [333435]. Currently, it is unknown if this effect exists in BRAFV600E-positive thyroid cancer patients. To address this question in an experimental setting, Roelli et al. [10] compared the response of thyroid cancer to the RAF inhibitor PLX4720 in Tg-CreERT2/BrafCA/Pik3caH1047R mice to that of Tg-CreERT2/BrafCA mice, demonstrating that activation of the PI3K pathway promoted resistance of the BRAFV600E-induced cancers to the RAF inhibitor. In addition, PLX4720 induced paradoxical activation of the MAPK pathway and stimulated tumor growth in Tg-CreERT2/BrafCA/Pik3caH1047R but not Tg-CreERT2/BrafCA mice. Combining the PI3K inhibitor GDC-0941 with PLX4720 restored inhibition of the MAPK pathway by the RAF inhibitor, along with suppressing the PI3K pathway, which produced a marked antitumor response [10]. Alternatively, ElMokh et al. [9] demonstrated a potent anti-tumor response in Tg-CreERT2/BrafCA/Pik3caH1047R mice treated with the MEK inhibitor, which was modestly enhanced by the addition of GDC-0941.
- Progression of tumors in the Tg-CreERT2/BrafCA/Pik3caH1047R and Tg-CreERT2/BrafCA/Ptenflox/flox mice to ATCs confirms the conclusion from human genomic data that activation of the PI3K pathway promotes progression of BRAFV600E thyroid cancers. The observation that Tg-CreERT2/BrafCA/Pik3caH1047R thyroid are resistant to RAF inhibitors suggests that BRAFV600E-positive human thyroid cancer with activation of the PI3K pathway would also be resistant. Conversely, the marked anti-tumor response to combined RAF and PI3K inhibitors in mice suggests this drug combination maybe effective in the equivalent human thyroid cancers. However, clinical trials in other cancers combining MAPK and PI3K kinase pathway inhibitors found significant toxicity [36] that will likely have to be addressed (i.e., isoform selective inhibitors or schedule optimization) for this to be a viable option for long-term thyroid cancer treatment. PI3K activation was demonstrated to induce resistance to RAF inhibitors. It remains to be determined if resistance induced by activation of the PI3K pathway would extend to the RAF/MEK inhibitor (i.e., dabrafenib/trametinib) combination that recently received U.S. Food and Drug Administration (FDA)-approval for treatment of BRAFV600E ATCs, and whether addition of PI3K inhibitor would improve the efficacy of this combination.
THYROID CANCER PROGRESSION AND PI3K PATHWAY
- As shown in Fig. 1, approximately 59% BRAFV600E-driven ATCs harbor alterations in TP53, but this combination is very rarely seen in other histotypes of thyroid cancers. Moreover, in the few PTCs identified with alterations in TP53, these mutations were subclonal [1]. These results strongly support inactivation of TP53 as an important step in progression of BRAFV600E PTCs to ATCs. This is supported by three different studies that demonstrated mice with inactivation of transformation related protein 53 (Trp53) and thyroid-specific expression of BrafV600E developed ATCs.
- McFadden et al. [21] was the first to report that the combination of BRAFV600E and inactivation of Trp53 promotes progression to ATCs. To demonstrate this, they crossed thyroid peroxidase promoter driven cre/estrogen receptor ligand binding domain fusion (TPO-CreERT2)/BrafCA/+ with Trp53f/f or Trp53LSL-R270H/f mice. The resulting TPO-CreERT2/BrafCA/+/Trp53LSL-R270H/F and TPO-CreERT2/BrafCA/+/Trp53f/f mice (referred to as TB270/FL and TBP, respectively) had tamoxifen-inducible, thyroid-specific knock-in of BrafV600E and either global expression of mutant Trp53 (Trp53LSL-R270H) or thyroid-specific deletion of exon 2–10 of Trp53 (Trp53f/f). Compared to TPO-CreERT2/Braf-CA/+, the tamoxifen-treated TPO-CreERT2/BrafCA/+/Trp53LSL-R270H/+ showed accelerated tumor growth, shorter survival, and histologic features associated with poor prognosis in human PTC [21]. In addition, 50% of the tumors in older animals showed progression to PDTC or ATC, which was not observed in TPO-CreERT2/BrafCA/+ mice. Homozygous inactivation of Trp53 further accelerated progression to ATC, which was accompanied by a decrease in survival. The authors report a modest but significant decrease in survival of tamoxifen-treated TB270/FL compared to TBP; however, the study was not designed to control for other factors that may contribute to this difference (i.e., tamoxifen dose, animal background).
- Zou et al. [24] created mice with thyroid-specific knock-in of BrafV600E and global loss of Trp53 to test the hypothesis that TSH promotes Braf-driven thyroid cancers by suppressing Trp53 and overcoming senescence induced by expression of BrafV600E. To this end, they crossed TPO-Cre, LSL-BrafV600E, and Trp53−/− mice to generate the TPO-Cre, LSL-BrafV600E/Trp53−/− (TPO-BrafV600E-Trp53+/−) and TPO-Cre/LSL-BrafV600E/Trp53+/− (TPO-BrafV600E-Trp53−/−) triple transgenic mice. Both mouse lines developed ATCs, with the TPO-BrafV600E-Trp53+/− having a delay in time to progression of 2 to 3 months.
- Knauf et al. [8] created mice with thyroid-specific homozygous loss of Trp53 and dox-inducible expression of BRAFV600E to explore mechanisms of acquired resistance of ATCs to BRAF inhibition. To generate this line, they crossed the following lines TPO-Cre, LSL-reverse tetracycline transcription activator-internal ribosomal entry site green fluorescent protein (LSL-rtTAires-GFP), tetracycline resistant operator-MYC proto oncogene tagged BRAFV600E (tetO-mycBRAFV600E), and Trp53f/f to generate the quadruple transgenic line, TPO-Cre/LSL-rtTAiresGFP/tetO-mycBRAFV600E/Trp53f/f (BRAF/p53). Induction of oncogenic BRAF in 4- to 8-week-old BRAF/p53 mice for 8 weeks produced ATCs in 50% of the mice that rapidly progressed, requiring euthanasia of all ATC bearing mice within 3 weeks of tumor detection.
- These three mouse models have differences in ATC latency, which is likely in part due to timing of oncogene activation and Trp53 loss. The ATCs in all three models were very similar to human ATCs, with histological analysis showing pleomorphic giant cells and spindle cells, high mitotic rate, and local invasion. In humans, BRAFV600E ATCs often coexist with a PTC component, which is seen in both TBP and TPO-BrafV600E-Trp53−/− mice. By contrast, a PTC component was not observed in BRAF/p53 mice. The reason for this difference is unclear, but a possible explanation is the timing of genetic changes. In TBP mice, loss of p53 and activation of Braf occur simultaneously in adults, whereas in BRAF/p53 animals, thyroid-specific Trp53 inactivation occurs late in embryonic development, followed by expression of oncogenic protein when the animals are adults. Similar to human ATCs, in all models, ATC bearing mice had a short survival and lung metastasis were frequently found in both TBP (19%) and BRAF/p53 (50%) mice [821]. Frequency of lung metastasis was not investigated in the TPO-BrafV600E-Trp53−/− mice.
- In humans, RAF inhibitors have shown efficacy in BRAFV600E ATC, which is further improved by combining it with MEK inhibitors [1112]. Indeed, the combination of RAF and MEK inhibitors dabrafenib and trametinib has recently been FDA-approved for the treatment of BRAFV600E positive ATCs [11]. In TBP mice, the RAF inhibitor PLX4720 induced a partial response in the PTC-bearing thyroid gland, but the ATC component continued to grow. There was also a modest extension of survival with the RAF inhibitor with all animals progressing within 35 days after starting treatment. In BRAF/p53 mice, PLX4720 did not have an effect, and cell lines generated from TPO-BrafV600E-Trp53−/− mice were resistant to RAF inhibitors. As in humans, treating murine ATCs in both TBP and BRAF/p53 with RAF and MEK inhibitor combination produced a more potent inhibition of the MAPK pathway and marked tumor regression that improved survival. Nevertheless, more than half of the TBP animals succumbed to the disease after approximately 70 days of treatment. Time to progression of BRAF/p53 mice treated with CH5126766 was not determined, but given that after genetic inhibition of BRAFV600E,which produced a near complete tumor regression, nearly all animals recurred [8], it is reasonable to think that mice treated with CH5126766 would also progress.
- In melanoma patients, multiple mechanisms of acquired resistance to RAF inhibitors as well as RAF/MEK inhibitor have been identified (reviewed in [37383940]). The majority of these reactivated the MAPK pathway. Mechanisms of aquired resistance include overexpression of CRAF or COT1, BRAFV600E amplification, activating mutations in NRAS, KRAS, MEK1, or AKT1 [3435414243], BRAFV600E alternative splicing, activation of phosphatidylinositol-3-OH kinase or increased expression of receptor tyrosine kinases. Other than a single case report identifying a NRASQ61K in BRAFV600E-positive PTC with acquired resistance to vemurafenib [44], there is no information on the mechanisms of acquired resistance of BRAF-mutant thyroid cancers to RAF kinase inhibition. Knauf et al. [8] investigated mechanisms of acquired resistance to genetic inhibition of BrafV600E in the BRAF/p53 mouse model. Transcriptome analysis demonstrated that reactivation of the MAPK pathway was common to all recurrences which retained sensitivity to the MEK/RAF inhibitor CH5126766. Whole exome sequencing identified recurrent focal amplifications of Met in 45% of the recurrent tumors as well as an activating mutation of Hras [8]. The Met-amplified recurrences overexpressed the receptor as well as its ligand hepatocyte growth factor (Hgf), and were exquisitely sensitive to MET kinase inhibitors, which was required for activation of the MAPK pathway [8]. While it remains to be demonstrated in human thyroid cancers, it seems likely that acquired resistance of BRAFV600E-positive thyroid cancers to RAF and RAF/MEK inhibitors will center around the reactivation of the MAPK pathway as seen in melanomas [37383940], which likely explains the improved response of ATCs to RAF/MEK inhibitor combination compared to RAF inhibitors alone [1112]. It will be important to determine events driving acquired resistance to RAF and RAF/MEK inhibitors in humans, especially for the latter, which is now FDA-approved in BRAFV600E-positive ATCs.
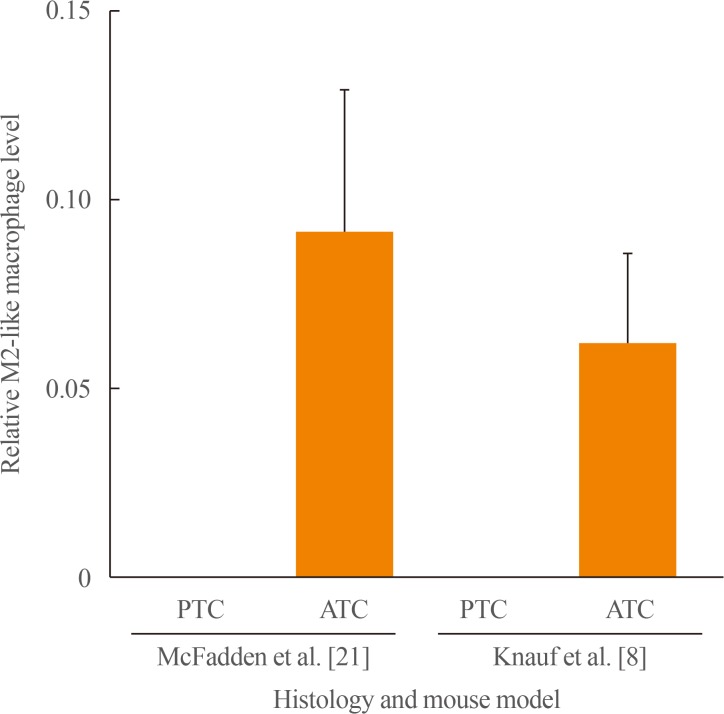
- The ATCs in the BRAF/p53 mice are heavily infiltrated with macrophages [8], which are almost universally found in their human counterparts [45]. Macrophage infiltration was not investigated in the ATCs from TBP or TPO-BrafV600E-Trp53−/− mice. However, immune deconvolution of transcriptomic data with CIBERSORT [46] demonstrated a similar increase in macrophages of ATCs from TBP to those from BRAF/p53 mice (Fig. 2). Macrophages have been suggested to promote tumor progression by stimulating angiogenesis [4748] and lymphangiogenesis, enhancing chemoresistance [495051], promoting distant metastases [5253545556], and creating an immune suppressive microenvironment. In human thyroid cancers, macrophage infiltration is associated with decreased survival in both PDTCs and ATCs [4557]. Depletion of macrophages in mouse models of BRAF-induced PTC attenuated tumor formation and restored a thyroid follicular architecture [58]. The effect of macrophage depletion on ATCs has yet to be investigated in either mouse models or human trials. Results in other cancers suggest that depletion of macrophages alone is unlikely to have profound or sustained anti-tumor effects but is likely to improve the efficacy of chemo-, targeted or immuno-therapies. The recent demonstration that inhibition of PI3Kγ promotes a change in macrophage polarization from M2-like to M1-like, which is associated with a markedly improved response to immune checkpoint blockade [5960], suggest altering macrophage polarization may be an effective alternative to macrophage depletion. As ATCs are frequently infiltrated with both exhausted T-cells [616263] and macrophages [4564], this combination might have efficacy in this aggressive disease.
- Human ATCs have been demonstrated to also be infiltrated with programmed cell death 1 (PD1)-positive T-cells, programmed death ligand 1 (PD-L1)-expressing macrophages, and possibly myeloid derived suppressor cells (MDSCs). Expression of PD1 and PD-L1 has been found to be associated with a positive response to anti-PD1/PD-L1 immunotherapies, whereas infiltration of the immune suppressive MDSCs and macrophages would be expected to promote resistance to immune therapies. Currently it has not been reported whether the mouse models of ATC also replicate the T-cell infiltrate, MDSC, PD1, or PD-L1 positivity of human ATCs. In the age of immunotherapy, it will be essential to expand our knowledge of the tumor microenvironment of human and mouse ATCs to guide investigators on how to best use the mouse models to understand the effects of the complex tumor microenvironment in ATCs on the response to immune therapies and improve the efficacy of immune therapies in advanced thyroid cancers.
INACTIVATION OF TRP53
- Genomic analysis of thyroid cancers at different stages of progression has identified several strong candidates for promoting progression of BRAFV600E-driven PTCs to ATCs (Table 1, Fig. 1). Of these, activation of Pik3ca and loss of Trp53 have been demonstrated to be sufficient to drive thyroid cancer progression in mouse models (Table 2). Probably due to its genetic simplicity, there is an excellent concordance between the alterations driving BRAFV600E-mutant progression in thyroid cancer patients and the murine models created to mimic these genetic events. This strongly suggests that results from genomic studies in large cohorts of advanced thyroid cancer patients should inform future approaches involving genetically engineered mouse model research. It also attests to the efficacy of these in vivo models to become essential tools to test pharmacological combinations that can be ultimately tested in appropriate clinical trials.
CONCLUSIONS
-
Acknowledgements
- The authors would like to thank James Fagin for his insightful discussion. This work was supported by NIH grants RO1-CA50706, RO1-CA72597, P50-CA72012, P30-CA008748, the Lefkovsky Family Foundation, the Linn Family Fund, the Society of Memorial Sloan Kettering and the Byrne fund.
ACKNOWLEDGMENTS
-
CONFLICTS OF INTEREST: No potential conflict of interest relevant to this article was reported.
Article information
- 1. Cancer Genome Atlas Research Network. Integrated genomic characterization of papillary thyroid carcinoma. Cell 2014;159:676–690. ArticlePubMedPMC
- 2. Kunstman JW, Juhlin CC, Goh G, Brown TC, Stenman A, Healy JM, et al. Characterization of the mutational landscape of anaplastic thyroid cancer via whole-exome sequencing. Hum Mol Genet 2015;24:2318–2329. ArticlePubMedPMCPDF
- 3. Ibrahimpasic T, Xu B, Landa I, Dogan S, Middha S, Seshan V, et al. Genomic alterations in fatal forms of non-anaplastic thyroid cancer: identification of MED12 and RBM10 as novel thyroid cancer genes associated with tumor virulence. Clin Cancer Res 2017;23:5970–5980. ArticlePubMedPMC
- 4. Landa I, Ibrahimpasic T, Boucai L, Sinha R, Knauf JA, Shah RH, et al. Genomic and transcriptomic hallmarks of poorly differentiated and anaplastic thyroid cancers. J Clin Invest 2016;126:1052–1066. ArticlePubMedPMC
- 5. Pozdeyev N, Gay LM, Sokol ES, Hartmaier R, Deaver KE, Davis S, et al. Genetic analysis of 779 advanced differentiated and anaplastic thyroid cancers. Clin Cancer Res 2018;24:3059–3068. ArticlePubMedPMC
- 6. Fagin JA, Wells SA Jr. Biologic and clinical perspectives on thyroid cancer. N Engl J Med 2016;375:2307.ArticlePMC
- 7. Leboeuf R, Baumgartner JE, Benezra M, Malaguarnera R, Solit D, Pratilas CA, et al. BRAFV600E mutation is associated with preferential sensitivity to mitogen-activated protein kinase kinase inhibition in thyroid cancer cell lines. J Clin Endocrinol Metab 2008;93:2194–2201. ArticlePubMedPMCPDF
- 8. Knauf JA, Luckett KA, Chen KY, Voza F, Socci ND, Ghossein R, et al. Hgf/Met activation mediates resistance to BRAF inhibition in murine anaplastic thyroid cancers. J Clin Invest 2018;128:4086–4097. ArticlePubMedPMC
- 9. ElMokh O, Ruffieux-Daidie D, Roelli MA, Stooss A, Phillips WA, Gertsch J, et al. Combined MEK and Pi3'-kinase inhibition reveals synergy in targeting thyroid cancer in vitro and in vivo. Oncotarget 2017;8:24604–24620. ArticlePubMedPMC
- 10. Roelli MA, Ruffieux-Daidie D, Stooss A, ElMokh O, Phillips WA, Dettmer MS, et al. PIK3CA(H1047R)-induced paradoxical ERK activation results in resistance to BRAF (V600E) specific inhibitors in BRAF(V600E) PIK3CA (H1047R) double mutant thyroid tumors. Oncotarget 2017;8:103207–103222. ArticlePubMedPMC
- 11. Hyman DM, Puzanov I, Subbiah V, Faris JE, Chau I, Blay JY, et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med 2015;373:726–736. ArticlePubMedPMC
- 12. Subbiah V, Kreitman RJ, Wainberg ZA, Cho JY, Schellens JHM, Soria JC, et al. Dabrafenib and trametinib treatment in patients with locally advanced or metastatic BRAF V600-mutant anaplastic thyroid cancer. J Clin Oncol 2018;36:7–13. ArticlePubMed
- 13. Knauf JA, Ma X, Smith EP, Zhang L, Mitsutake N, Liao XH, et al. Targeted expression of BRAFV600E in thyroid cells of transgenic mice results in papillary thyroid cancers that undergo dedifferentiation. Cancer Res 2005;65:4238–4245. ArticlePubMed
- 14. Rusinek D, Swierniak M, Chmielik E, Kowal M, Kowalska M, Cyplinska R, et al. BRAFV600E-associated gene expression profile: early changes in the transcriptome, based on a transgenic mouse model of papillary thyroid carcinoma. PLoS One 2015;10:e0143688. ArticlePubMedPMC
- 15. Franco AT, Malaguarnera R, Refetoff S, Liao XH, Lundsmith E, Kimura S, et al. Thyrotrophin receptor signaling dependence of Braf-induced thyroid tumor initiation in mice. Proc Natl Acad Sci U S A 2011;108:1615–1620. ArticlePubMedPMC
- 16. Charles RP, Iezza G, Amendola E, Dankort D, McMahon M. Mutationally activated BRAF(V600E) elicits papillary thyroid cancer in the adult mouse. Cancer Res 2011;71:3863–3871. ArticlePubMedPMC
- 17. Shimamura M, Shibusawa N, Kurashige T, Mussazhanova Z, Matsuzaki H, Nakashima M, et al. Mouse models of sporadic thyroid cancer derived from BRAFV600E alone or in combination with PTEN haploinsufficiency under physiologic TSH levels. PLoS One 2018;13:e0201365. ArticlePubMedPMC
- 18. Chakravarty D, Santos E, Ryder M, Knauf JA, Liao XH, West BL, et al. Small-molecule MAPK inhibitors restore radioiodine incorporation in mouse thyroid cancers with conditional BRAF activation. J Clin Invest 2011;121:4700–4711. ArticlePubMedPMC
- 19. Nagarajah J, Le M, Knauf JA, Ferrandino G, Montero-Conde C, Pillarsetty N, et al. Sustained ERK inhibition maximizes responses of BrafV600E thyroid cancers to radioiodine. J Clin Invest 2016;126:4119–4124. ArticlePubMedPMC
- 20. Charles RP, Silva J, Iezza G, Phillips WA, McMahon M. Activating BRAF and PIK3CA mutations cooperate to promote anaplastic thyroid carcinogenesis. Mol Cancer Res 2014;12:979–986. ArticlePubMedPMC
- 21. McFadden DG, Vernon A, Santiago PM, Martinez-McFaline R, Bhutkar A, Crowley DM, et al. p53 constrains progression to anaplastic thyroid carcinoma in a Braf-mutant mouse model of papillary thyroid cancer. Proc Natl Acad Sci U S A 2014;111:E1600–E1609. ArticlePubMedPMC
- 22. Knauf JA, Sartor MA, Medvedovic M, Lundsmith E, Ryder M, Salzano M, et al. Progression of BRAF-induced thyroid cancer is associated with epithelial-mesenchymal transition requiring concomitant MAP kinase and TGFβ signaling. Oncogene 2011;30:3153–3162. ArticlePubMedPMCPDF
- 23. Shimamura M, Nakahara M, Orim F, Kurashige T, Mitsutake N, Nakashima M, et al. Postnatal expression of BRAFV600E does not induce thyroid cancer in mouse models of thyroid papillary carcinoma. Endocrinology 2013;154:4423–4430. ArticlePubMedPDF
- 24. Zou M, Baitei EY, Al-Rijjal RA, Parhar RS, Al-Mohanna FA, Kimura S, et al. TSH overcomes Braf(V600E)-induced senescence to promote tumor progression via downregulation of p53 expression in papillary thyroid cancer. Oncogene 2016;35:1909–1918. ArticlePubMedPDF
- 25. Chen Y, Sadow PM, Suh H, Lee KE, Choi JY, Suh YJ, et al. BRAF(V600E) is correlated with recurrence of papillary thyroid microcarcinoma: a systematic review, multi-institutional primary data analysis, and meta-analysis. Thyroid 2016;26:248–255. ArticlePubMed
- 26. Cetani F, Tonacchera M, Vassart G. Differential effects of NaCl concentration on the constitutive activity of the thyrotropin and the luteinizing hormone/chorionic gonadotropin receptors. FEBS Lett 1996;378:27–31. ArticlePubMed
- 27. Mitsutake N, Knauf JA, Mitsutake S, Mesa C Jr, Zhang L, Fagin JA. Conditional BRAFV600E expression induces DNA synthesis, apoptosis, dedifferentiation, and chromosomal instability in thyroid PCCL3 cells. Cancer Res 2005;65:2465–2473. ArticlePubMed
- 28. Liu D, Hu S, Hou P, Jiang D, Condouris S, Xing M. Suppression of BRAF/MEK/MAP kinase pathway restores expression of iodide-metabolizing genes in thyroid cells expressing the V600E BRAF mutant. Clin Cancer Res 2007;13:1341–1349. ArticlePubMed
- 29. Ho AL, Grewal RK, Leboeuf R, Sherman EJ, Pfister DG, Deandreis D, et al. Selumetinib-enhanced radioiodine uptake in advanced thyroid cancer. N Engl J Med 2013;368:623–632. ArticlePubMedPMC
- 30. Ishii N, Harada N, Joseph EW, Ohara K, Miura T, Sakamoto H, et al. Enhanced inhibition of ERK signaling by a novel allosteric MEK inhibitor, CH5126766, that suppresses feedback reactivation of RAF activity. Cancer Res 2013;73:4050–4060. ArticlePubMedPMC
- 31. Rothenberg SM, Daniels GH, Wirth LJ. Redifferentiation of iodine-refractory BRAF V600E-mutant metastatic papillary thyroid cancer with dabrafenib-response. Clin Cancer Res 2015;21:5640–5641. ArticlePubMed
- 32. Dunn LA, Sherman EJ, Baxi SS, Tchekmedyian V, Grewal RK, Larson SM, et al. Vemurafenib redifferentiation of BRAF mutant, RAI-refractory thyroid cancers. J Clin Endocrinol Metab 2018 9 25 [Epub]. ArticlePDF
- 33. Shi H, Hong A, Kong X, Koya RC, Song C, Moriceau G, et al. A novel AKT1 mutant amplifies an adaptive melanoma response to BRAF inhibition. Cancer Discov 2014;4:69–79. ArticlePubMed
- 34. Shi H, Hugo W, Kong X, Hong A, Koya RC, Moriceau G, et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov 2014;4:80–93. ArticlePubMed
- 35. Hugo W, Shi H, Sun L, Piva M, Song C, Kong X, et al. Non-genomic and immune evolution of melanoma acquiring MAPKi resistance. Cell 2015;162:1271–1285. ArticlePubMedPMC
- 36. Schram AM, Gandhi L, Mita MM, Damstrup L, Campana F, Hidalgo M, et al. A phase Ib dose-escalation and expansion study of the oral MEK inhibitor pimasertib and PI3K/MTOR inhibitor voxtalisib in patients with advanced solid tumours. Br J Cancer 2018;119:1471–1476. ArticlePubMedPMCPDF
- 37. Kakadia S, Yarlagadda N, Awad R, Kundranda M, Niu J, Naraev B, et al. Mechanisms of resistance to BRAF and MEK inhibitors and clinical update of US Food and Drug Administration-approved targeted therapy in advanced melanoma. Onco Targets Ther 2018;11:7095–7107. ArticlePubMedPMC
- 38. Welsh SJ, Rizos H, Scolyer RA, Long GV. Resistance to combination BRAF and MEK inhibition in metastatic melanoma: Where to next? Eur J Cancer 2016;62:76–85. ArticlePubMed
- 39. Lito P, Rosen N, Solit DB. Tumor adaptation and resistance to RAF inhibitors. Nat Med 2013;19:1401–1409. ArticlePubMedPDF
- 40. Sullivan RJ, Flaherty KT. Resistance to BRAF-targeted therapy in melanoma. Eur J Cancer 2013;49:1297–1304. ArticlePubMed
- 41. Shi H, Moriceau G, Kong X, Lee MK, Lee H, Koya RC, et al. Melanoma whole-exome sequencing identifies (V600E) B-RAF amplification-mediated acquired B-RAF inhibitor resistance. Nat Commun 2012;3:724ArticlePubMedPMC
- 42. Johnson DB, Menzies AM, Zimmer L, Eroglu Z, Ye F, Zhao S, et al. Acquired BRAF inhibitor resistance: a multicenter meta-analysis of the spectrum and frequencies, clinical behaviour, and phenotypic associations of resistance mechanisms. Eur J Cancer 2015;51:2792–2799. ArticlePubMedPMC
- 43. Rizos H, Menzies AM, Pupo GM, Carlino MS, Fung C, Hyman J, et al. BRAF inhibitor resistance mechanisms in metastatic melanoma: spectrum and clinical impact. Clin Cancer Res 2014;20:1965–1977. ArticlePubMed
- 44. Ofir Dovrat T, Sokol E, Frampton G, Shachar E, Pelles S, Geva R, et al. Unusually long-term responses to vemurafenib in BRAF V600E mutated colon and thyroid cancers followed by the development of rare RAS activating mutations. Cancer Biol Ther 2018;19:871–874. ArticlePubMedPMC
- 45. Ryder M, Ghossein RA, Ricarte-Filho JC, Knauf JA, Fagin JA. Increased density of tumor-associated macrophages is associated with decreased survival in advanced thyroid cancer. Endocr Relat Cancer 2008;15:1069–1074. ArticlePubMedPMC
- 46. Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods 2015;12:453–457. ArticlePubMedPMCPDF
- 47. Lin EY, Pollard JW. Tumor-associated macrophages press the angiogenic switch in breast cancer. Cancer Res 2007;67:5064–5066. ArticlePubMed
- 48. Laoui D, Van Overmeire E, Di Conza G, Aldeni C, Keirsse J, Morias Y, et al. Tumor hypoxia does not drive differentiation of tumor-associated macrophages but rather fine-tunes the M2-like macrophage population. Cancer Res 2014;74:24–30. ArticlePubMed
- 49. Schoppmann SF, Birner P, Stockl J, Kalt R, Ullrich R, Caucig C, et al. Tumor-associated macrophages express lymphatic endothelial growth factors and are related to peritumoral lymphangiogenesis. Am J Pathol 2002;161:947–956. ArticlePubMedPMC
- 50. Werchau S, Toberer F, Enk A, Dammann R, Helmbold P. Merkel cell carcinoma induces lymphatic microvessel formation. J Am Acad Dermatol 2012;67:215–225. ArticlePubMed
- 51. Weizman N, Krelin Y, Shabtay-Orbach A, Amit M, Binenbaum Y, Wong RJ, et al. Macrophages mediate gemcitabine resistance of pancreatic adenocarcinoma by upregulating cytidine deaminase. Oncogene 2014;33:3812–3819. ArticlePubMedPDF
- 52. Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell 2010;141:39–51. ArticlePubMedPMC
- 53. Gocheva V, Wang HW, Gadea BB, Shree T, Hunter KE, Garfall AL, et al. IL-4 induces cathepsin protease activity in tumor-associated macrophages to promote cancer growth and invasion. Genes Dev 2010;24:241–255. ArticlePubMedPMC
- 54. Wyckoff JB, Wang Y, Lin EY, Li JF, Goswami S, Stanley ER, et al. Direct visualization of macrophage-assisted tumor cell intravasation in mammary tumors. Cancer Res 2007;67:2649–2656. ArticlePubMed
- 55. Liu CY, Xu JY, Shi XY, Huang W, Ruan TY, Xie P, et al. M2-polarized tumor-associated macrophages promoted epithelial-mesenchymal transition in pancreatic cancer cells, partially through TLR4/IL-10 signaling pathway. Lab Invest 2013;93:844–854. ArticlePubMedPDF
- 56. Wang D, Sun H, Wei J, Cen B, DuBois RN. CXCL1 is critical for premetastatic niche formation and metastasis in colorectal cancer. Cancer Res 2017;77:3655–3665. ArticlePubMedPMC
- 57. Kim DI, Kim E, Kim YA, Cho SW, Lim JA, Park YJ. Macrophage densities correlated with CXC chemokine receptor 4 expression and related with poor survival in anaplastic thyroid cancer. Endocrinol Metab (Seoul) 2016;31:469–475. ArticlePubMedPMC
- 58. Ryder M, Gild M, Hohl TM, Pamer E, Knauf J, Ghossein R, et al. Genetic and pharmacological targeting of CSF-1/CSF-1R inhibits tumor-associated macrophages and impairs BRAF-induced thyroid cancer progression. PLoS One 2013;8:e54302. ArticlePubMedPMC
- 59. Kaneda MM, Messer KS, Ralainirina N, Li H, Leem CJ, Gorjestani S, et al. PI3Kγ is a molecular switch that controls immune suppression. Nature 2016;539:437–442. ArticlePubMedPMCPDF
- 60. De Henau O, Rausch M, Winkler D, Campesato LF, Liu C, Cymerman DH, et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kγ in myeloid cells. Nature 2016;539:443–447. ArticlePubMedPMCPDF
- 61. Chintakuntlawar AV, Rumilla KM, Smith CY, Jenkins SM, Foote RL, Kasperbauer JL, et al. Expression of PD-1 and PD-L1 in anaplastic thyroid cancer patients treated with multimodal therapy: results from a retrospective study. J Clin Endocrinol Metab 2017;102:1943–1950. ArticlePubMedPDF
- 62. Bastman JJ, Serracino HS, Zhu Y, Koenig MR, Mateescu V, Sams SB, et al. Tumor-infiltrating T cells and the PD-1 checkpoint pathway in advanced differentiated and anaplastic thyroid cancer. J Clin Endocrinol Metab 2016;101:2863–2873. ArticlePubMedPMC
- 63. Zwaenepoel K, Jacobs J, De Meulenaere A, Silence K, Smits E, Siozopoulou V, et al. CD70 and PD-L1 in anaplastic thyroid cancer: promising targets for immunotherapy. Histopathology 2017;71:357–365. ArticlePubMed
- 64. Caillou B, Talbot M, Weyemi U, Pioche-Durieu C, Al Ghuzlan A, Bidart JM, et al. Tumor-associated macrophages (TAMs) form an interconnected cellular supportive network in anaplastic thyroid carcinoma. PLoS One 2011;6:e22567. ArticlePubMedPMC
References
Genetic alterations in human BRAFV600E-driven thyroid cancers. Oncoprint showing the most frequent mutations identified in BRAFV600E-mutant of papillary thyroid carcinomas from the The Cancer Genome Atlas study (PTC-TCGA, n=235, left), poorly-differentiated thyroid carcinomas (PDTCs, n=32, middle), and anaplastic thyroid cancers (ATCs, n=85, right). Mutation data compiled from TCGA [1], Kunstman et al. [2], Landa et al. [4], Ibrahimpasic et al. [3], Pozdeyev et al. [5], and Memorial Sloan Kettering Cancer Center Clinical Runs, as of November 1st, 2018. TP53, tumor protein p53; TERT, telomerase reverse transcriptase; PIK3CA, phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha; SWI/SNF, mating-type switching/sucrose non-fermenting complex; NF2, neurofibromin 2; CDKN2A, cyclin dependent kinase inhibitor 2A; RBM10, RNA binding motif protein 10.

M2-like macrophage infiltration in oncogenic BRAF-driven mouse anaplastic thyroid cancers (ATCs). Immune deconvolution using CIBERSORT [46] was performed on transcriptomic data from the following mouse models: McFadden et al. [21] (GSE55933) TPO-CreERT2/Braf (papillary thyroid carcinoma [PTC]) and TPO-CreERT2/BrafCA/Trp53f/f (ATC). Knauf et al. [8] (GSE118022) PTC, LSL-Braf/TPO-Cre: ATC, TPO-Cre/LSL-rtTAiresGFP/tetO-myc-BRAFV600E/Trp53f/f. Bars show the relative level of M2-like macrophages in the indicated model and cancer histologies. TPO-CreERT2, thyroid peroxidase driven cre/estrogen receptor ligand binding domain fusion; BrafCA, Cre-activated BrafV600E allele; Trp53, transformation related protein 53; LSL, lox-stop-lox; rtTAiresGFP, reverse tetracycline transcription activator-internal ribosomal entry site-green fluorescent protein; tetO-mycBRAFV600E, tetracycline resistant operator-MYC proto oncogene tagged BRAFV600E.
The Number of Mutant Samples for Each of the Genes and Pathways for the Indicated Histology Displayed on the Oncoprint (Fig. 1) and Contingency Analysis of These Genetic Events in PTC vs. PDTC vs. ATC
PTC, papillary thyroid carcinoma; PDTC, poorly-differentiated thyroid carcinoma; ATC, anaplastic thyroid cancer; TP53, tumor protein p53; TERT, telomerase reverse transcriptase; PIK3CA, phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha; SWI/SNF, mating-type switching/sucrose non-fermenting complex; NF2, neurofibromin 2; CDKN2A, cyclin dependent kinase inhibitor 2A; RBM10, RNA binding motif protein 10.
aFisher's exact test significant P value.
Mouse Models of BRAFV600E-Driven Thyroid Cancers
| Mouse alleles | Reference | Pros | Cons |
|---|---|---|---|
| PTC | |||
| TG-BRAF | [1322] | Single transgene | BRAFV600E overexpressed and not under control of endogenous promoter |
| LSL-BrafV600E/TPO-Cre | [1519] | BrafV600E under control of endogenous promoter; short latency with near 100% PTC penetrance | Oncoprotein induction at a single time point |
| TG-CreERT2/BrafCA | [16] | BrafV600E under control of endogenous promoter; flexibility on when oncoprotein is induced | Long latency; leaky Cre activity |
| TPO-CreERT2/BrafCA | [21] | BrafV600E under control of endogenous promoter; flexibility on when oncoprotein is induced | Long latency |
| BrafCA/TPO-Cre | [17] | BrafV600E under control of endogenous promoter; short latency with near 100% PTC penetrance | Oncoprotein induction at a single time point |
| TG-BRAFV600E | [14] | Single transgene | BRAFV600E overexpressed and not under control of endogenous promoter |
| TG-rtTA/tetO-mycBraf | [18] | Short latency, flexibility on when oncoprotein is induced; can regulate expression of oncoprotein | BRAFV600E overexpressed and not under control of endogenous promoter |
| TPO-Cre/LNL-BRAFV600E | [23] | Flexibility on when oncoprotein is induced | No tumor formation in absence of elevated TSH; BRAFV600E overexpressed and not under control of endogenous promoter |
| PDTC/ATC | |||
| TG-CreERT2/BrafCA/Ptenf/f | [20] | BrafV600E under control of endogenous promoter; flexibility on when oncoprotein is induced | BrafV600E induction and loss of Pten simultaneous which doesn't mimic common human scenario |
| TG-CreERT2/BrafCA/Pik3ca | [20] | BrafV600E under control of endogenous promoter; flexibility on when oncoprotein is induced | BrafV600E and Pik3ca induction simultaneous which doesn't mimic common human scenario |
| TPO-CreERT2/BrafCA/Trp53f/f | [21] | BrafV600E under control of endogenous promoter; flexibility on when oncoprotein is induced | BrafV600E induction and loss of Trp53 simultaneous which doesn't mimic common human scenario |
| TPO-Cre/LSL-rtTAiresGFP/tetO-mycBRAFV600E/Trp53f/f | [2] | Short latency, flexibility on when oncoprotein is induced; can regulate expression of oncoprotein | BRAFV600E not under control of endogenous promoter |
| TPO-Cre/LSL-BrafV600E/Trp53−/– | [24] | BrafV600E under control of endogenous promoter; short latency | Oncoprotein induction at a single time point; other tumors likely due to germline Trp53 inactivation |
PTC, papillary thyroid carcinoma; TG, thyroglobulin; LSL, lox-stop-lox; TPO, thyroid peroxidase; CreERT2, Cre/estrogen receptor ligand binding domain fusion; BrafCA, Cre-activated BrafV600E allele; rtTA, reverse tetracycline transcription activator; tetO-mycBRAFV600E, tetracycline resistant operator-MYC proto oncogene tagged BRAFV600E; LNL, loxP-neoR-loxP; TSH, thyroid-stimulating hormone; PDTC, poorly-differentiated thyroid carcinoma; ATC, anaplastic thyroid cancer; Pten, phosphatase and tensin homolog; Pik3ca, phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha; Trp53, transformation related protein 53; rtTAiresGFP, reverse tetracycline transcription activator-internal ribosomal entry site–green fluorescent protein.
Figure & Data
References
Citations
- Strategies to investigate migration and metastases in thyroid cancer
Daniel M. Chopyk, Priya H. Dedhia
Current Opinion in Endocrine and Metabolic Research.2024; 34: 100502. CrossRef - Comparative efficiency of differential diagnostic methods for the identification of BRAF V600E gene mutation in papillary thyroid cancer (Review)
Qian Liu, Xue Jiang, Wenling Tu, Lina Liu, Ying Huang, Yuxiao Xia, Xuliang Xia, Yuhong Shi
Experimental and Therapeutic Medicine.2024;[Epub] CrossRef - Mouse Models to Examine Differentiated Thyroid Cancer Pathogenesis: Recent Updates
Hye Choi, Kwangsoon Kim
International Journal of Molecular Sciences.2023; 24(13): 11138. CrossRef - Mechanistic Insights of Thyroid Cancer Progression
Luis Javier Leandro-García, Iñigo Landa
Endocrinology.2023;[Epub] CrossRef - Anaplastic thyroid cancer: Pathogenesis, prognostic factors and genetic landscape (Review)
Abdul-Mohsen Alhejaily, Omar Alhuzim, Yazeed Alwelaie
Molecular and Clinical Oncology.2023;[Epub] CrossRef - Tissue architecture delineates field cancerization in BRAFV600E-induced tumor development
Elin Schoultz, Ellen Johansson, Carmen Moccia, Iva Jakubikova, Naveen Ravi, Shawn Liang, Therese Carlsson, Mikael Montelius, Konrad Patyra, Jukka Kero, Kajsa Paulsson, Henrik Fagman, Martin O. Bergo, Mikael Nilsson
Disease Models & Mechanisms.2022;[Epub] CrossRef - BRAFV600E Expression in Thyrocytes Causes Recruitment of Immunosuppressive STABILIN-1 Macrophages
Catherine Spourquet, Ophélie Delcorte, Pascale Lemoine, Nicolas Dauguet, Axelle Loriot, Younes Achouri, Maija Hollmén, Sirpa Jalkanen, François Huaux, Sophie Lucas, Pierre Van Meerkeeck, Jeffrey A. Knauf, James A. Fagin, Chantal Dessy, Michel Mourad, Patr
Cancers.2022; 14(19): 4687. CrossRef - Positive BRAFV600E mutation of primary tumor influences radioiodine avidity but not prognosis of papillary thyroid cancer with lung metastases
Shuhui Huang, Mengfang Qi, Tian Tian, Hongyuan Dai, Yuan Tang, Rui Huang
Frontiers in Endocrinology.2022;[Epub] CrossRef - Preclinical Models of Follicular Cell-Derived Thyroid Cancer: An Overview from Cancer Cell Lines to Mouse Models
Min Ji Jeon, Bryan R. Haugen
Endocrinology and Metabolism.2022; 37(6): 830. CrossRef - An Animal Model Further Uncovers the Role of Mutant Braf during Papillary Thyroid Cancer Development
Bernd Koelsch, Sarah Theurer, Magdalena Staniszewska, Jacqueline Heupel, Amelie Koch, Svenja Mergener, Franziska Walk, Christine Fischer, Andrea Kutritz, Kurt W. Schmid, Andrea Kindler-Röhrborn
The American Journal of Pathology.2020; 190(3): 702. CrossRef - Intratumoral Genetic Heterogeneity in Papillary Thyroid Cancer: Occurrence and Clinical Significance
Laura Fugazzola, Marina Muzza, Gabriele Pogliaghi, Mario Vitale
Cancers.2020; 12(2): 383. CrossRef - The Aryl Hydrocarbon Receptor Is Expressed in Thyroid Carcinoma and Appears to Mediate Epithelial-Mesenchymal-Transition
Sonia Moretti, Nicole Nucci, Elisa Menicali, Silvia Morelli, Vittorio Bini, Renato Colella, Martina Mandarano, Angelo Sidoni, Efisio Puxeddu
Cancers.2020; 12(1): 145. CrossRef
 PubReader
PubReader ePub Link
ePub Link Cite
Cite


