Articles
- Page Path
- HOME > Endocrinol Metab > Volume 34(3); 2019 > Article
-
Review ArticleRadioactive Iodine-Refractory Differentiated Thyroid Cancer and Redifferentiation Therapy
-
Jierui Liu1,2,3
 , Yanqing Liu1,2, Yansong Lin1,2,3, Jun Liang4
, Yanqing Liu1,2, Yansong Lin1,2,3, Jun Liang4 -
Endocrinology and Metabolism 2019;34(3):215-225.
DOI: https://doi.org/10.3803/EnM.2019.34.3.215
Published online: September 26, 2019
1Department of Nuclear Medicine, Peking Union Medical College Hospital, Beijing, China.
2Beijing Key Laboratory of Molecular Targeted Diagnosis and Therapy in Nuclear Medicine, Beijing, China.
3Department of Oncology, The Affiliated Hospital of Qingdao University, Qingdao, China.
4Department of Oncology, Peking University International Hospital, Beijing, China.
- Corresponding author: Yansong Lin. Department of Nuclear Medicine, Peking Union Medical College Hospital, No. 1 Shuaifuyuan, Wangfujing St, Dongcheng, Beijing 100730, China. Tel: +86-10-69155610, Fax: +86-10-69155610, linys@pumch.cn
- Corresponding author: Jun Liang. Department of Oncology, Peking University International Hospital, No.1 Life Park St., Zhongguancun Life Science Park, Haidian, Beijing 102206, China. Tel: +86-10-69006624, Fax: +86-10-69006082, liangjun1959@aliyun.com
Copyright © 2019 Korean Endocrine Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
- The retained functionality of the sodium iodide symporter (NIS) expressed in differentiated thyroid cancer (DTC) cells allows the further utilization of post-surgical radioactive iodine (RAI) therapy, which is an effective treatment for reducing the risk of recurrence, and even the mortality, of DTC. Whereas, the dedifferentiation of DTC could influence the expression of functional NIS, thereby reducing the efficacy of RAI therapy in advanced DTC. Genetic alternations (such as BRAF and the rearranged during transfection [RET]/papillary thyroid cancer [PTC] rearrangement) have been widely reported to be prominently responsible for the onset, progression, and dedifferentiation of PTC, mainly through activating the mitogen-activated protein kinase (MAPK) and phosphoinositide 3-kinase (PI3K) signaling cascades. These genetic alternations have been suggested to associate with the reduced expression of iodide-handling genes in thyroid cancer, especially the NIS gene, disabling iodine uptake and causing resistance to RAI therapy. Recently, novel and promising approaches aiming at various targets have been attempted to restore the expression of these iodine-metabolizing genes and enhance iodine uptake through in vitro studies and studies of RAI-refractory (RAIR)-DTC patients. In this review, we discuss the regulation of NIS, known mechanisms of dedifferentiation including the MAPK and PI3K pathways, and the current status of redifferentiation therapy for RAIR-DTC patients.
- Thyroid cancer has emerged as a striking health issue over recent decades because of its gradually increasing incidence worldwide. The global incidence of thyroid cancer is 6.7 per 100,000, and the number of newly diagnosed likely cases in China has exceeded 190,000 (194,232 cases) [1]. Papillary thyroid cancer (PTC), follicular thyroid cancer, and Hürthle cell cancer are derived from follicular cells; thus, they are collectively characterized as differentiated thyroid cancer (DTC), and account for more than 90% of all thyroid malignancies [23]. Although most DTC cases have a quite favorable prognosis after standard therapeutic approaches, including surgery, selective radioactive iodine (RAI) therapy, and thyroid stimulating hormone (TSH) suppressive therapy, the risk of local recurrence and distant metastasis may be up to 20% and 10%, respectively. Among these patients, two-thirds show initial or gradual loss of the ability of iodine uptake due to the dysfunction, and even loss, of sodium iodide symporter (NIS) expression in the basal membrane, indicating a status of dedifferentiation known as RAI-refractory DTC (RAIR-DTC), which is of major clinical concern because its 10-year survival rate is less than 10% [4].
- Genetic alterations are the fundamental drivers for the tumorigenesis and pathogenesis of thyroid cancer, aberrantly activating the mitogen-activated protein kinase (MAPK) and phosphoinositide 3-kinase (PI3K) pathways [56]. These alternations are also known to be associated with the silencing of various thyroid iodine-metabolizing genes, especially solute carrier family 5 member 5 (SLC5A5), which encodes NIS, thus resulting in the failure of RAI therapy [789]. Multiple agents have been investigated in attempts to restore NIS expression and to enhance RAI uptake in RAIR-DTC patients. This review aimed to summarize the regulation of NIS, the possible mechanism of NIS downregulation, and the potential of redifferentiation therapy for RAIR-DTC.
INTRODUCTION
- Iodide, as an essential component, is utilized by follicular cells to synthesize thyroid hormone in the normal thyroid. As it is expressed on the basal membrane, NIS provides the physiological basis for the active transport of iodide into the follicular cells of the thyroid [10]. DTC cells can retain similar functions to those of follicular cells, such as iodine uptake and iodination [1112], which allows RAI therapy to be the mainstay for the treatment of intermediate and high-risk DTC after surgery [13]. To destroy residual or potential subclinical lesions, RAI therapy could improve disease-specific survival and progression-free survival [14]. Hence, the function or expression of NIS in DTC cells is crucial for the efficacy of RAI therapy in such patients.
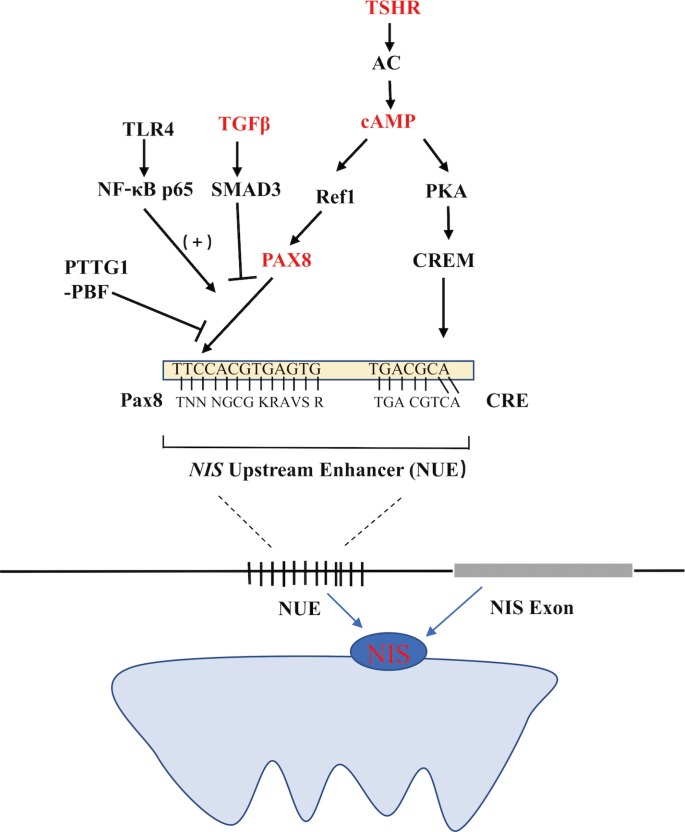
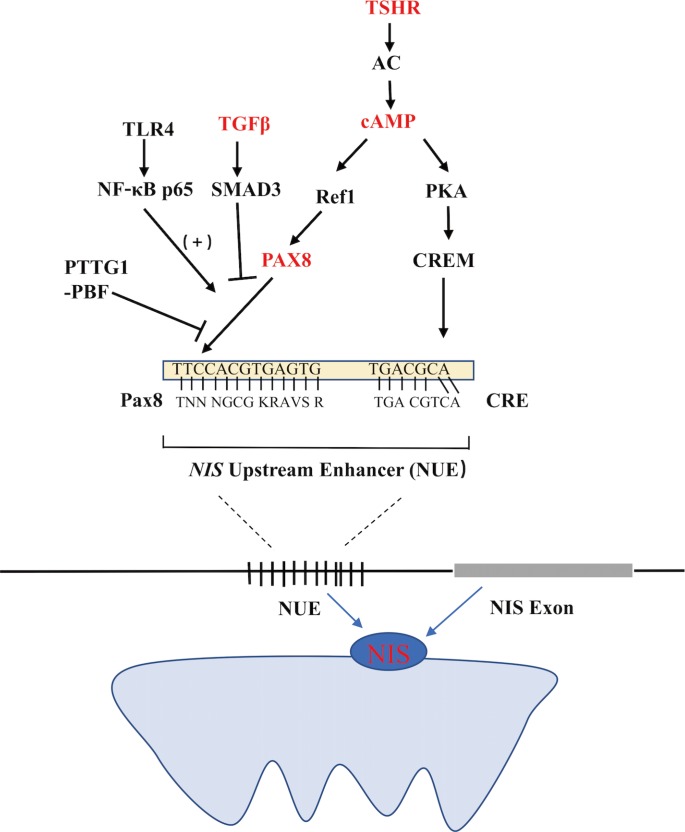
- Functional NIS expression can be regulated at both the transcriptional and post-translational levels. As the predominant regulator of NIS expression, TSH is primarily involved at the translational level. After the binding of TSH with the TSH receptor, adenylyl cyclase is stimulated through the Gs-protein, which in turn increases the expression of cyclic adenosine monophosphate (cAMP). cAMP then induces NIS transcription by activating several signaling pathways that could stimulate the NIS upstream enhancer (NUE). It is now known that the human NUE consists of paired box gene-8 (PAX8; thyroid-specific transcription factor) binding site and cAMP-response element like site, both of which are important for the integrated activity of the NUE [15]. As illustrated in Fig. 1, cAMP can stimulate the NUE through both protein kinase A (PKA)-independent and -dependent pathways. Through redox effector factor-1 (Ref-1), the PKA-independent pathway, PAX8 is subsequently stimulated to bind to the NUE, leading to the activation of NUE [15161718], and this pathway plays a key role in the differentiation of the thyroid [19]. Through the PKA-dependent pathway, the activated PKA could phosphorylate the cAMP-responsive element modulator, enhancing NUE activity [2021].
- TSH-independent mechanisms also regulate NIS expression, which mainly include three pathways influencing the binding of PAX8 to NUE. First, in the transforming growth factor β (TGFβ)-SMAD signaling pathway, TGFβ activates the downstream Smad3 and subsequently inhibits the binding of PAX8 to NUE, significantly decreasing NIS mRNA expression in thyroid cells [916]. Second, in the Toll-like receptor (TLR)-nuclear factor κ-light-chain-enhancer of activated B cells (NF-κB) signaling pathway, the TLR activates the downstream NF-κB, which further interacts with PAX8, activating NIS transcription via the NUE [2223]. Third, in the pituitary tumor-transforming gene-1 product (PTTG1)-binding factor (PBF) complex, the PTTG1 and the PBF could interfere with the binding of PAX8 to the NUE, thus suppressing the expression of NIS (Fig. 1) [242526]. Saez et al. [26] reported that increased PTTG1 expression could reduce the efficacy of RAI therapy in thyroid cancer.
- Concerning post-translational regulation, abundant NIS expression may mis-localize in the intracellular compartment rather than the cell membrane [27]. This abnormal membrane targeting of NIS could disable iodide transport and result in the reduced uptake and accumulation of RAI in thyroid cancer cells, inducing the probable failure of RAI therapy in a subset of DTC.
PHYSIOLOGICAL REGULATION OF NIS EXPRESSION
- MAPK/ERK pathway
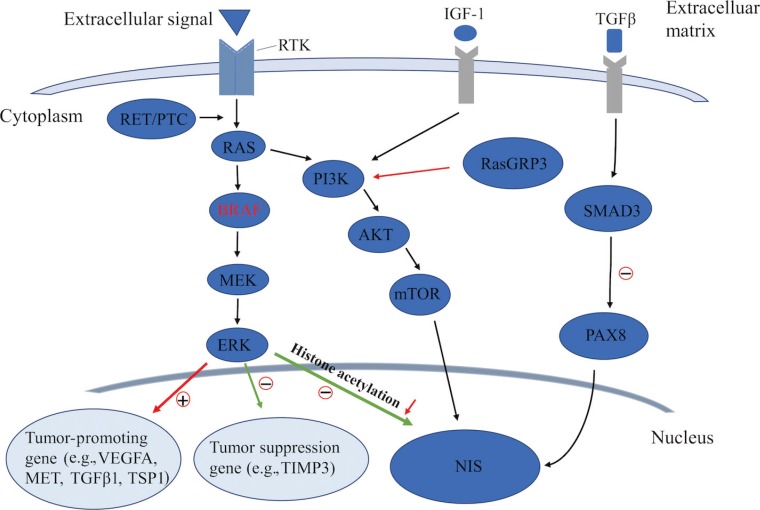
- The MAPK pathway has been well recognized and established in the regulation of cell proliferation, dedifferentiation, and survival [282930], particularly for PTC. Among the signal molecules of this pathway, BRAF mutations and the rearranged during transfection (RET)/PTC rearrangement are frequently detected in PTC, which often exhibit as exclusive mutations in such patients [53132]. The BRAFV600E mutation, as a prominently prevalent oncogene, is critical for the initiation and/or progression of PTC through aberrantly activating the MAPK signaling pathway, which can downregulate the expression of thyroid iodide-handling genes, especially NIS, and thus induce the dedifferentiation of PTC [2833343536]. Increasing evidence has demonstrated a strong association between the BRAFV600E mutation and the loss of RAI-avidity in PTC [37383940], which could provide a reasonable explanation for the failure of RAI therapy in BRAFV600E-mutant PTC.
- As an important epigenetic event, histone acetylation plays a fundamental role in the regulation of gene transcription [4142]. Through aberrant activation of the MAPK pathway, histone acetylation at the promoter of the gene encoding NIS could be downregulated, which is considered as one of the key molecular events involving the aberrant silencing of thyroid iodide-handling genes [543444546]. Meanwhile, the BRAFV600E mutation can also upregulate the expression of tumor-promoting genes (e.g., vascular endothelial growth factor A [VEGFA], mesenchymal to epithelial transition factor [MET], TGFβ1, and thrombospondin 1 [TSP1]) [934474849] and downregulate the expression of tumor suppressor genes (e.g., tissue inhibitor of metalloproteinases 3 [TIMP3], solute carrier family 5 member 8 [SLC5A8], and death-associated protein kinase 1 [DAPK1]) [50], which are important constituents of the tumor microenvironment. It is noteworthy that the autocrine TGFβ loop could play a role in aberrant NIS expression. Riesco-Eizaguirre et al. [9] and Costamagna et al. [16] reported that the BRAFV600E mutation could induce the secretion of TGFβ, which subsequently stimulated SMAD3 and impaired PAX8, causing a decrease of NIS expression. As this process was independent of the MAPK pathway, their result undoubtedly indicates that TGFβ could be considered as a candidate therapeutic target for the restoration of NIS expression in patients with advanced DTC (Fig. 2).
- It was recently recognized that telomerase reverse transcriptase (TERT) promoter (TERTp) mutations are particularly prevalent in aggressive thyroid cancers, especially BRAFV600E-mutant PTC, but are virtually undetectable in benign thyroid neoplasms [5152]. TERTp mutations were demonstrated to be associated with aggressive tumor behavior and poor prognosis in thyroid cancer [535455], and were also observed to be correlated with the reduction of RAI uptake in distant metastatic lesions of PTC [56], which suggested that TERTp mutations may play a role in the dedifferentiation of thyroid cancer. It was also reported that the duet alternation of BRAFV600E and TERTp mutations have a robust synergistic effect on the progression and poor clinical outcomes of PTC. Recently, Liu et al. [57] revealed that the BRAFV600E/MAPK pathway could phosphorylate and activate fructooligosaccharide, which in turn acted as a transcription factor to activate the GA binding protein transcription factor subunit beta (GABPB) promoter, increasing GABPB expression and leading to the formation of the GA binding protein transcription factor subunit alpha (GABPA)-GABPB complex, thus activating the mutant TERT promoter and upregulating TERT expression. The discovery of TERTp mutations and the genetic duet of coexisting mutations could afford promising molecular targets for the salvage therapy of RAIR-PTC.
- Concerning the RET/PTC rearrangement, evidence about its impact on the dedifferentiation of DTC remains limited. Trapasso et al. [58] and Wang et al. [59] reported that alternation of RET/PTC in the thyroid cell line could decrease the expression of thyroid differentiation markers, such as TSH receptor, TPO, NIS, and thyroglobulin. Furthermore, exogenous RET/PTC could significantly suppress the expression of PAX8 and the activity of PKA, leading to reduced NIS expression [858].
- PI3K/AKT pathway
- In addition to the MAPK pathway, the PI3K/AKT pathway also plays a fundamental role in controlling both cell proliferation and differentiation in DTC. In recent years, several genetic alterations activating the PI3K pathway have been identified in thyroid cancer, which was also shown to downregulate iodide-handling genes in thyroid cells [560]. Song et al. [61] recently reported that, also by this pathway, mutation of Ras guanyl releasing protein 3 (RasGRP3) decreased the expression of NIS and the TSH receptor in metastases of RAIR-DTC. Furthermore, the PI3K pathway could be activated by multiple growth factors such as insulin/insulin-like growth factor-1 (IGF-1) and epidermal growth factor [62]. Garcia and Santisteban [63] reported that IGF-1 could inhibit TSH-dependent NIS expression and reduce the iodide uptake in fisher rat thyroid cell line-5 (FRTL-5) rat thyroid cells through activating the PI3K/AKT pathway (Fig. 2). It has been shown that inhibition of the PI3K pathway by LY294002 could significantly increase the expression of NIS mRNA in rat thyroid cells and PTC cells, which, from another aspect, indicated the important role of the PI3K pathway in regulating NIS-mediated iodide accumulation in thyroid cancer [64]. The serine-threonine protein kinase mechanistic target of rapamycin (mTOR), which is located downstream of the PI3K/AKT pathway, has been identified as a regulator of cellular metabolism and proliferation. Souza et al. [65] reported that mTOR inhibition not only regulated cell survival, but also increased RAI uptake in both in vitro and in vivo studies.
KNOWN PATHWAYS DOWNREGULATING THE EXPRESSION OF NIS
- During the past decade, although great progress has been achieved in the treatment of RAIR-DTC with the application of multi-kinase inhibitors (MKIs), therapeutic options for these patients are still limited. As RAI resistance is predominantly due to the dedifferentiation of DTC, redifferentiation therapy followed by RAI therapy undoubtedly is a promising alternative option for RAIR-DTC patients. Several agents, including retinoic acid (RA) [66], peroxisome proliferator-activated receptor gamma (PPARγ) agonists [67], and histone deacetylase (HDAC) inhibitors [68], have been tried to modulate the NIS gene at the transcription level, but displayed limited clinical value in redifferentiation therapy for patients with RAIR-DTC. Recent studies using drugs that selectively inhibit the MAPK and PI3K pathways showed promising results for restoring the expression of the gene encoding NIS and improving the response to RAI therapy in RAIR-DTC, such as mitogen-activated extracellular signal-regulated kinase (MEK)/RAF inhibitors [6970], PI3K/mTOR inhibitors [64] and receptor tyrosine kinase (RTK) inhibitors [71], which were used to restore NIS expression by suppressing the signaling pathways in such patients.
- Agents modulating NIS at the gene transcriptional level
- RA is a biologically active metabolite of vitamin A that play key roles in cell differentiation and proliferation. RA has been used for redifferentiation treatment of thyroid cancer, exerting its effects via retinoid receptors, RA receptors (RAR), or retinoid X receptors (RXR). Studies have shown that NIS mRNA expression was upregulated by RA stimulation in human follicular thyroid carcinoma cell lines [72]. Several early small cohort studies showed that 40% to 50% of patients with RAIR-DTC experienced renewed RAI uptake after RA treatment [737475]. However, such promising results could not be repeated by subsequent studies, which disappointingly indicated that only 6% to 20% of patients showed increased RAI uptake after RA administration [667677]. A recent open-label, non-randomized phase II trial reported even more disappointing results, with only one patient (1/16) showing increased RAI uptake after RA administration [66].
- The PPARγ is a nuclear receptor which can regulate the proliferation and redifferentiation of the cell through forming a heterodimer with RXR [7879]. Pieces of evidence have shown that the rosiglitazone, a PPARγ agonist agent for the redifferentiation treatment of thyroid cancer, increased NIS mRNA expression and RAI uptake in thyroid cells [8081]. However, the results of a phase II clinical trial were disappointing, as 25% (5/20) of the patients displayed positive RAI uptake after rosiglitazone treatment, but no clinical response in long-term follow-up [67].
- HDAC is an enzyme that deacetylates histones, which could silence the expression of NIS in thyroid cancer [5]. HDAC inhibitors were found to increase the expression of NIS mRNA at the epigenetic level in an in vitro study [82]. Preclinical studies have shown renewed RAI uptake after treatment with various HDAC inhibitors, such as suberoylanilide hydroxamic acid (SAHA), depsipeptide (romidepsin), and valproic acid. Nevertheless, their independent significance in clinical trials was limited. Kelly et al. [83] reported that only one (1/3) advanced thyroid cancer patient showed increased RAI uptake after SAHA administration. In a phase I clinical trial of romidepsin, no positive RAI uptake was detected by RAI scintigraphy among 11 enrolled RAIR-DTC patients [84]. Furthermore, the results of a phase II clinical trial using romidepsin showed that only two patients experienced increased RAI uptake and no major response was observed in 20 patients with RAIR-DTC [68]. Nilubol et al. [85] performed a phase II clinical trial to evaluate the effect of valproic acid, without renewed RAI uptake.
- MAPK inhibitors
- The independent administration of retinoids, romidepsin, and rosiglitazone was shown to have limited clinical application in redifferentiation therapy for RAIR-DTC. With the development of molecular and cellular biology, multiple novel targets have been revealed, which could afford new options for the redifferentiation therapy of RAIR-DTC. Evidence has shown that agents selectively inhibiting the MAPK pathway, such as BRAF or MEK inhibitors, could induce thyroid gene expression and restore RAI uptake in thyroid cancer cells [4386]. The results of a clinical trial performed by Ho et al. [69] revealed that, after treatment with selumetinib, a selective MEK inhibitor, 124I uptake was increased in RAIR-PTC patients, which indicated the restoration of NIS expression in these patients. Meanwhile, it was interesting that the efficacy of selumetinib was better in patients with an NRAS mutation than those with the BRAFV600E mutation [69]. Furthermore, the prior phase II clinical trial of selumetinib seemed to suggest that BRAFV600E mutant patients exhibited longer median progression-free survival than patients with BRAF wild-type tumors [87]. This suggested a potential relationship between the therapeutic efficacy of selumetinib and genetic alterations. However, it was disappointing that the subsequent phase III trial of selumetinib failed to reconfirm its effect in the restoration of RAI uptake, which suggested that further studies are needed to explore the efficacy of MEK inhibitors in the redifferentiation of RAIR-DTC.
- Another phase II clinical trial showed that dabrafenib, a BRAF inhibitor, increased RAI uptake in 60% (6/10) of BRAF-mutant RAIR-DTC patients, with two of the six achieving partial response after subsequent RAI therapy [88], which surpassed selumetinib in the treatment of BRAF-mutated RAIR-DTC. Vemurafenib, another BRAF inhibitor, could also restore RAI uptake in a subset of BRAF-mutant RAIR-DTC patients, which was likely due to the upregulation of thyroid-specific gene expression via inhibition of the MAPK pathway [70]. Earlier studies showed that HDAC inhibitors (such as SAHA) alone could induce the expression of NIS and faint RAI uptake in thyroid cancer cells [8990]; gratifyingly, a recent study reported that combined treatment with an HDAC inhibitor and a MAPK inhibitor (dabrafenib and selumetinib) showed a robust redifferentiation effect in BRAFV600E-mutated thyroid cancer cells [91]. This result suggests that the combined administration of HDAC and/or MAPK inhibitors might be a promising choice to improve the efficacy of redifferentiation therapy in RAIR-DTC patients. Furthermore, Nagarajah et al. [92] reported that extracellular regulated protein kinase (ERK) inhibitors could significantly increase the accumulation of 124I in BRAFV600E-mutant PTC cells, indicating that ERK inhibitors may be a potential candidate for redifferentiation therapy in BRAFV600E-mutant PTC.
- PI3K inhibitors
- As mentioned above, the aberrant activation of the PI3K/AKT/mTOR pathway could downregulate NIS expression [5], indicating that this pathway may be a potential therapeutic target for the redifferentiation therapy of RAIR-DTC. LY294002, a PI3K inhibitor, significantly upregulated the expression of NIS mRNA and improved iodide uptake through induction of PAX8 in DTC cell lines [6493]. Moreover, the inhibition of AKT also exhibited an increase of iodide uptake by mediating the expression of NIS in thyroid cells [93]. Plantinga et al. [94] reported that an mTOR inhibitor could induce iodine uptake through increasing thyroid transcription factor 1 (TTF1) expression in an in vitro study. Nevertheless, findings have not been reported from several in vivo studies that were initiated to evaluate the changes of iodine uptake by suppressing the PI3K pathway in RAIR-DTC patients. Hence, further studies might be necessary to elucidate the impact of PI3K/AKT/mTOR pathway inhibitors on the redifferentiation of RAIR-DTC.
- RTK inhibitors
- RTKs, such as vascular endothelial growth factor receptor (VEGFR), RET, platelet-derived growth factor receptors (PDGFRs), and human epidermal growth factor receptor (HER), are also crucial molecules inducing the dedifferentiation of DTC. By aberrantly causing the MAPK pathway to rebound, RTKs, such as HER3, could also lead to resistance to MKIs in RAIR-DTC patients [95]. Agents targeting RTKs have been recently investigated for the redifferentiation and salvage therapy of MKI-resistant RAIR-DTC. Cheng et al. [71] reported that combination therapy of BRAF/MEK inhibitors (dabrafenib/selumetinib) with an HER inhibitor (lapatinib) could upregulate NIS expression and suppress the MAPK pathway without a rebound phenomenon, and the corresponding phase I trial is now underway. The above-mentioned results may afford us a new prospective for the redifferentiation therapy of RAIR-DTC, as well as alternative salvage therapy for MKI-resistant advanced thyroid cancer.
REDIFFERENTIATION THERAPY FOR RAIR-DTC
- The expression of NIS allows RAI therapy to be a highly effective management strategy for DTC, especially in metastatic DTC patients. Genetic alternations could reduce the expression of NIS and lead to the dedifferentiation of DTC, mainly through activation of the MAPK and PI3K pathways in thyroid cancer, causing a RAIR state that represents a life-threatening clinical situation. Efforts have been made to restore NIS expression and enhance RAI avidity in RAIR-DTC. Agents for dedifferentiation therapy at the transcriptional level have yielded limited clinical impact in clinical trials. Kinase inhibitors targeting the MAPK or PI3K pathways have shown promising effects in redifferentiation therapy and shed light on future combination therapy between either kinase inhibitors with different targets or kinase inhibitors and RAI in the management of RAIR-DTC.
CONCLUSIONS
-
Acknowledgements
- This review was supported by the National Natural Sciences Foundation of China (81771875 and 81571714), the Medicine and Technology Innovation Project of the Chinese Academy of Medical Science (grant number 2016-12M-2-006), and the Thyroid Study Group Project of the Asia Oceania Research Initiative Network (AORIN).
ACKNOWLEDGMENTS
-
CONFLICTS OF INTEREST: No potential conflict of interest relevant to this article was reported.
Article information
- 1. Ferlay J, Colombet M, Soerjomataram I, Mathers C, Parkin DM, Pineros M, et al. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int J Cancer 2019;144:1941–1953. ArticlePubMed
- 2. Mao Y, Xing M. Recent incidences and differential trends of thyroid cancer in the USA. Endocr Relat Cancer 2016;23:313–322. ArticlePubMedPMC
- 3. Lim H, Devesa SS, Sosa JA, Check D, Kitahara CM. Trends in thyroid cancer incidence and mortality in the United States, 1974-2013. JAMA 2017;317:1338–1348. ArticlePubMedPMC
- 4. Durante C, Haddy N, Baudin E, Leboulleux S, Hartl D, Travagli JP, et al. Long-term outcome of 444 patients with distant metastases from papillary and follicular thyroid carcinoma: benefits and limits of radioiodine therapy. J Clin Endocrinol Metab 2006;91:2892–2899. ArticlePubMedPDF
- 5. Xing M. Molecular pathogenesis and mechanisms of thyroid cancer. Nat Rev Cancer 2013;13:184–199. ArticlePubMedPMCPDF
- 6. Zaballos MA, Santisteban P. Key signaling pathways in thyroid cancer. J Endocrinol 2017;235:R43–R61. ArticlePubMed
- 7. Xing M. BRAF mutation in thyroid cancer. Endocr Relat Cancer 2005;12:245–262. ArticlePubMed
- 8. Venkateswaran A, Marsee DK, Green SH, Jhiang SM. Forskolin, 8-Br-3′,5′-cyclic adenosine 5′-monophosphate, and catalytic protein kinase A expression in the nucleus increase radioiodide uptake and sodium/iodide symporter protein levels in RET/PTC1-expressing cells. J Clin Endocrinol Metab 2004;89:6168–6172. ArticlePubMed
- 9. Riesco-Eizaguirre G, Rodriguez I, De la Vieja A, Costamagna E, Carrasco N, Nistal M, et al. The BRAFV600E oncogene induces transforming growth factor beta secretion leading to sodium iodide symporter repression and increased malignancy in thyroid cancer. Cancer Res 2009;69:8317–8325. ArticlePubMed
- 10. Riesco-Eizaguirre G, Santisteban P. A perspective view of sodium iodide symporter research and its clinical implications. Eur J Endocrinol 2006;155:495–512. ArticlePubMed
- 11. Castro MR, Bergert ER, Goellner JR, Hay ID, Morris JC. Immunohistochemical analysis of sodium iodide symporter expression in metastatic differentiated thyroid cancer: correlation with radioiodine uptake. J Clin Endocrinol Metab 2001;86:5627–5632. ArticlePubMedPDF
- 12. Wapnir IL, van de Rijn M, Nowels K, Amenta PS, Walton K, Montgomery K, et al. Immunohistochemical profile of the sodium/iodide symporter in thyroid, breast, and other carcinomas using high density tissue microarrays and conventional sections. J Clin Endocrinol Metab 2003;88:1880–1888. ArticlePubMed
- 13. American Thyroid Association (ATA) Guidelines Taskforce on Thyroid Nodules and Differentiated Thyroid Cancer. Cooper DS, Doherty GM, Haugen BR, Kloos RT, Lee SL, et al. Revised American Thyroid Association management guidelines for patients with thyroid nodules and differentiated thyroid cancer. Thyroid 2009;19:1167–1214. ArticlePubMed
- 14. Tuttle RM, Ahuja S, Avram AM, Bernet VJ, Bourguet P, Daniels GH, et al. Controversies, consensus, and collaboration in the use of (131)I therapy in differentiated thyroid cancer: a joint statement from the American Thyroid Association, the European Association of Nuclear Medicine, the Society of Nuclear Medicine and Molecular Imaging, and the European Thyroid Association. Thyroid 2019;29:461–470. ArticlePubMed
- 15. Taki K, Kogai T, Kanamoto Y, Hershman JM, Brent GA. A thyroid-specific far-upstream enhancer in the human sodium/iodide symporter gene requires Pax-8 binding and cyclic adenosine 3′,5′-monophosphate response element-like sequence binding proteins for full activity and is differentially regulated in normal and thyroid cancer cells. Mol Endocrinol 2002;16:2266–2282. ArticlePubMedPDF
- 16. Costamagna E, Garcia B, Santisteban P. The functional interaction between the paired domain transcription factor Pax8 and Smad3 is involved in transforming growth factor-beta repression of the sodium/iodide symporter gene. J Biol Chem 2004;279:3439–3446. ArticlePubMed
- 17. Kambe F, Nomura Y, Okamoto T, Seo H. Redox regulation of thyroid-transcription factors, Pax-8 and TTF-1, is involved in their increased DNA-binding activities by thyrotropin in rat thyroid FRTL-5 cells. Mol Endocrinol 1996;10:801–812. ArticlePubMed
- 18. Tell G, Pellizzari L, Cimarosti D, Pucillo C, Damante G. Ref-1 controls pax-8 DNA-binding activity. Biochem Biophys Res Commun 1998;252:178–183. ArticlePubMed
- 19. Mansouri A, Chowdhury K, Gruss P. Follicular cells of the thyroid gland require Pax8 gene function. Nat Genet 1998;19:87–90. ArticlePubMedPDF
- 20. Chun JT, Di Dato V, D'Andrea B, Zannini M, Di Lauro R. The CRE-like element inside the 5′-upstream region of the rat sodium/iodide symporter gene interacts with diverse classes of b-Zip molecules that regulate transcriptional activities through strong synergy with Pax-8. Mol Endocrinol 2004;18:2817–2829. ArticlePubMed
- 21. Fenton MS, Marion KM, Hershman JM. Identification of cyclic adenosine 3′,5′-monophosphate response element modulator as an activator of the human sodium/iodide symporter upstream enhancer. Endocrinology 2008;149:2592–2606. ArticlePubMedPMCPDF
- 22. Nicola JP, Velez ML, Lucero AM, Fozzatti L, Pellizas CG, Masini-Repiso AM. Functional toll-like receptor 4 conferring lipopolysaccharide responsiveness is expressed in thyroid cells. Endocrinology 2009;150:500–508. ArticlePubMedPDF
- 23. Nicola JP, Nazar M, Mascanfroni ID, Pellizas CG, Masini-Repiso AM. NF-kappaB p65 subunit mediates lipopolysaccharide-induced Na(+)/I(−) symporter gene expression by involving functional interaction with the paired domain transcription factor Pax8. Mol Endocrinol 2010;24:1846–1862. ArticlePubMedPMCPDF
- 24. Boelaert K, Smith VE, Stratford AL, Kogai T, Tannahill LA, Watkinson JC, et al. PTTG and PBF repress the human sodium iodide symporter. Oncogene 2007;26:4344–4356. ArticlePubMedPDF
- 25. Stratford AL, Boelaert K, Tannahill LA, Kim DS, Warfield A, Eggo MC, et al. Pituitary tumor transforming gene binding factor: a novel transforming gene in thyroid tumorigenesis. J Clin Endocrinol Metab 2005;90:4341–4349. ArticlePubMedPDF
- 26. Saez C, Martinez-Brocca MA, Castilla C, Soto A, Navarro E, Tortolero M, et al. Prognostic significance of human pituitary tumor-transforming gene immunohistochemical expression in differentiated thyroid cancer. J Clin Endocrinol Metab 2006;91:1404–1409. ArticlePubMed
- 27. Kogai T, Brent GA. The sodium iodide symporter (NIS): regulation and approaches to targeting for cancer therapeutics. Pharmacol Ther 2012;135:355–370. ArticlePubMedPMC
- 28. Xing M. BRAF mutation in papillary thyroid cancer: pathogenic role, molecular bases, and clinical implications. Endocr Rev 2007;28:742–762. ArticlePubMedPDF
- 29. Cohen Y, Xing M, Mambo E, Guo Z, Wu G, Trink B, et al. BRAF mutation in papillary thyroid carcinoma. J Natl Cancer Inst 2003;95:625–627. ArticlePubMedPDF
- 30. Liu D, Liu Z, Condouris S, Xing M. BRAF V600E maintains proliferation, transformation, and tumorigenicity of BRAF-mutant papillary thyroid cancer cells. J Clin Endocrinol Metab 2007;92:2264–2271. ArticlePubMedPMCPDF
- 31. Kimura ET, Nikiforova MN, Zhu Z, Knauf JA, Nikiforov YE, Fagin JA. High prevalence of BRAF mutations in thyroid cancer: genetic evidence for constitutive activation of the RET/PTC-RAS-BRAF signaling pathway in papillary thyroid carcinoma. Cancer Res 2003;63:1454–1457. PubMed
- 32. Soares P, Trovisco V, Rocha AS, Lima J, Castro P, Preto A, et al. BRAF mutations and RET/PTC rearrangements are alternative events in the etiopathogenesis of PTC. Oncogene 2003;22:4578–4580. ArticlePubMedPDF
- 33. Kim TH, Park YJ, Lim JA, Ahn HY, Lee EK, Lee YJ, et al. The association of the BRAF(V600E) mutation with prognostic factors and poor clinical outcome in papillary thyroid cancer: a meta-analysis. Cancer 2012;118:1764–1773. ArticlePubMed
- 34. Giordano TJ, Kuick R, Thomas DG, Misek DE, Vinco M, Sanders D, et al. Molecular classification of papillary thyroid carcinoma: distinct BRAF, RAS, and RET/PTC mutation-specific gene expression profiles discovered by DNA microarray analysis. Oncogene 2005;24:6646–6656. ArticlePubMedPDF
- 35. Riesco-Eizaguirre G, Gutierrez-Martinez P, Garcia-Cabezas MA, Nistal M, Santisteban P. The oncogene BRAF V600E is associated with a high risk of recurrence and less differentiated papillary thyroid carcinoma due to the impairment of Na+/I− targeting to the membrane. Endocr Relat Cancer 2006;13:257–269. ArticlePubMed
- 36. Mian C, Barollo S, Pennelli G, Pavan N, Rugge M, Pelizzo MR, et al. Molecular characteristics in papillary thyroid cancers (PTCs) with no 131I uptake. Clin Endocrinol (Oxf) 2008;68:108–116. ArticlePubMed
- 37. Xing M, Westra WH, Tufano RP, Cohen Y, Rosenbaum E, Rhoden KJ, et al. BRAF mutation predicts a poorer clinical prognosis for papillary thyroid cancer. J Clin Endocrinol Metab 2005;90:6373–6379. ArticlePubMed
- 38. Ricarte-Filho JC, Ryder M, Chitale DA, Rivera M, Heguy A, Ladanyi M, et al. Mutational profile of advanced primary and metastatic radioactive iodine-refractory thyroid cancers reveals distinct pathogenetic roles for BRAF, PIK3CA, and AKT1. Cancer Res 2009;69:4885–4893. ArticlePubMedPMC
- 39. Barollo S, Pennelli G, Vianello F, Watutantrige Fernando S, Negro I, Merante Boschin I, et al. BRAF in primary and recurrent papillary thyroid cancers: the relationship with (131)I and 2-[(18)F]fluoro-2-deoxy-D-glucose uptake ability. Eur J Endocrinol 2010;163:659–663. ArticlePubMed
- 40. Yang K, Wang H, Liang Z, Liang J, Li F, Lin Y. BRAFV600E mutation associated with non-radioiodine-avid status in distant metastatic papillary thyroid carcinoma. Clin Nucl Med 2014;39:675–679. ArticlePubMed
- 41. Barneda-Zahonero B, Parra M. Histone deacetylases and cancer. Mol Oncol 2012;6:579–589. ArticlePubMedPMC
- 42. Horikoshi M. Histone acetylation: from code to web and router via intrinsically disordered regions. Curr Pharm Des 2013;19:5019–5042. ArticlePubMed
- 43. Hou P, Bojdani E, Xing M. Induction of thyroid gene expression and radioiodine uptake in thyroid cancer cells by targeting major signaling pathways. J Clin Endocrinol Metab 2010;95:820–828. ArticlePubMedPDF
- 44. Durante C, Puxeddu E, Ferretti E, Morisi R, Moretti S, Bruno R, et al. BRAF mutations in papillary thyroid carcinomas inhibit genes involved in iodine metabolism. J Clin Endocrinol Metab 2007;92:2840–2843. ArticlePubMedPDF
- 45. Di Cristofaro J, Silvy M, Lanteaume A, Marcy M, Carayon P, De Micco C. Expression of tpo mRNA in thyroid tumors: quantitative PCR analysis and correlation with alterations of ret, Braf, ras and pax8 genes. Endocr Relat Cancer 2006;13:485–495. ArticlePubMed
- 46. Zhang Z, Liu D, Murugan AK, Liu Z, Xing M. Histone deacetylation of NIS promoter underlies BRAF V600E-promoted NIS silencing in thyroid cancer. Endocr Relat Cancer 2014;21:161–173. ArticlePubMedPMC
- 47. Jo YS, Li S, Song JH, Kwon KH, Lee JC, Rha SY, et al. Influence of the BRAF V600E mutation on expression of vascular endothelial growth factor in papillary thyroid cancer. J Clin Endocrinol Metab 2006;91:3667–3670. ArticlePubMedPDF
- 48. Nucera C, Porrello A, Antonello ZA, Mekel M, Nehs MA, Giordano TJ, et al. B-Raf(V600E) and thrombospondin-1 promote thyroid cancer progression. Proc Natl Acad Sci U S A 2010;107:10649–10654. ArticlePubMedPMC
- 49. Knauf JA, Sartor MA, Medvedovic M, Lundsmith E, Ryder M, Salzano M, et al. Progression of BRAF-induced thyroid cancer is associated with epithelial-mesenchymal transition requiring concomitant MAP kinase and TGFβ signaling. Oncogene 2011;30:3153–3162. ArticlePubMedPMCPDF
- 50. Hu S, Liu D, Tufano RP, Carson KA, Rosenbaum E, Cohen Y, et al. Association of aberrant methylation of tumor suppressor genes with tumor aggressiveness and BRAF mutation in papillary thyroid cancer. Int J Cancer 2006;119:2322–2329. ArticlePubMed
- 51. Liu R, Xing M. TERT promoter mutations in thyroid cancer. Endocr Relat Cancer 2016;23:R143–R155. ArticlePubMedPMC
- 52. Alzahrani AS, Alsaadi R, Murugan AK, Sadiq BB. TERT promoter mutations in thyroid cancer. Horm Cancer 2016;7:165–177. ArticlePubMedPMCPDF
- 53. Sohn SY, Park WY, Shin HT, Bae JS, Ki CS, Oh YL, et al. Highly concordant key genetic alterations in primary tumors and matched distant metastases in differentiated thyroid cancer. Thyroid 2016;26:672–682. ArticlePubMed
- 54. Bu R, Siraj AK, Divya SP, Kong Y, Parvathareddy SK, Al-Rasheed M, et al. Telomerase reverse transcriptase mutations are independent predictor of disease-free survival in Middle Eastern papillary thyroid cancer. Int J Cancer 2018;142:2028–2039. ArticlePubMed
- 55. Liu R, Bishop J, Zhu G, Zhang T, Ladenson PW, Xing M. Mortality risk stratification by combining BRAF V600E and TERT promoter mutations in papillary thyroid cancer: genetic duet of BRAF and TERT promoter mutations in thyroid cancer mortality. JAMA Oncol 2017;3:202–208. ArticlePubMed
- 56. Yang X, Li J, Li X, Liang Z, Gao W, Liang J, et al. TERT promoter mutation predicts radioiodine-refractory character in distant metastatic differentiated thyroid cancer. J Nucl Med 2017;58:258–265. ArticlePubMed
- 57. Liu R, Zhang T, Zhu G, Xing M. Regulation of mutant TERT by BRAF V600E/MAP kinase pathway through FOS/GABP in human cancer. Nat Commun 2018;9:579ArticlePubMedPMC
- 58. Trapasso F, Iuliano R, Chiefari E, Arturi F, Stella A, Filetti S, et al. Iodide symporter gene expression in normal and transformed rat thyroid cells. Eur J Endocrinol 1999;140:447–451. ArticlePubMed
- 59. Wang J, Knauf JA, Basu S, Puxeddu E, Kuroda H, Santoro M, et al. Conditional expression of RET/PTC induces a weak oncogenic drive in thyroid PCCL3 cells and inhibits thyrotropin action at multiple levels. Mol Endocrinol 2003;17:1425–1436. ArticlePubMed
- 60. Xing M. Genetic alterations in the phosphatidylinositol-3 kinase/Akt pathway in thyroid cancer. Thyroid 2010;20:697–706. ArticlePubMedPMC
- 61. Song J, Qiu W, Deng X, Qiu Z, Fan Y, Yang Z. A somatic mutation of RasGRP3 decreases Na(+)/I(−) symporter expression in metastases of radioactive iodine-refractory thyroid cancer by stimulating the Akt signaling pathway. Am J Cancer Res 2018;8:1847–1855. PubMedPMC
- 62. Kimura T, Van Keymeulen A, Golstein J, Fusco A, Dumont JE, Roger PP. Regulation of thyroid cell proliferation by TSH and other factors: a critical evaluation of in vitro models. Endocr Rev 2001;22:631–656. ArticlePubMedPDF
- 63. Garcia B, Santisteban P. PI3K is involved in the IGF-I inhibition of TSH-induced sodium/iodide symporter gene expression. Mol Endocrinol 2002;16:342–352. ArticlePubMed
- 64. Kogai T, Sajid-Crockett S, Newmarch LS, Liu YY, Brent GA. Phosphoinositide-3-kinase inhibition induces sodium/iodide symporter expression in rat thyroid cells and human papillary thyroid cancer cells. J Endocrinol 2008;199:243–252. ArticlePubMed
- 65. de Souza EC, Padron AS, Braga WM, de Andrade BM, Vaisman M, Nasciutti LE, et al. MTOR downregulates iodide uptake in thyrocytes. J Endocrinol 2010;206:113–120. ArticlePubMed
- 66. Short SC, Suovuori A, Cook G, Vivian G, Harmer C. A phase II study using retinoids as redifferentiation agents to increase iodine uptake in metastatic thyroid cancer. Clin Oncol (R Coll Radiol) 2004;16:569–574. ArticlePubMed
- 67. Kebebew E, Lindsay S, Clark OH, Woeber KA, Hawkins R, Greenspan FS. Results of rosiglitazone therapy in patients with thyroglobulin-positive and radioiodine-negative advanced differentiated thyroid cancer. Thyroid 2009;19:953–956. ArticlePubMed
- 68. Sherman EJ, Su YB, Lyall A, Schoder H, Fury MG, Ghossein RA, et al. Evaluation of romidepsin for clinical activity and radioactive iodine reuptake in radioactive iodine-refractory thyroid carcinoma. Thyroid 2013;23:593–599. ArticlePubMedPMC
- 69. Ho AL, Grewal RK, Leboeuf R, Sherman EJ, Pfister DG, Deandreis D, et al. Selumetinib-enhanced radioiodine uptake in advanced thyroid cancer. N Engl J Med 2013;368:623–632. ArticlePubMedPMC
- 70. Dunn LA, Sherman EJ, Baxi SS, Tchekmedyian V, Grewal RK, Larson SM, et al. Vemurafenib redifferentiation of BRAF mutant, RAI-refractory thyroid cancers. J Clin Endocrinol Metab 2019;104:1417–1428. ArticlePubMedPDF
- 71. Cheng L, Jin Y, Liu M, Ruan M, Chen L. HER inhibitor promotes BRAF/MEK inhibitor-induced redifferentiation in papillary thyroid cancer harboring BRAFV600E. Oncotarget 2017;8:19843–19854. ArticlePubMedPMC
- 72. Schmutzler C, Schmitt TL, Glaser F, Loos U, Kohrle J. The promoter of the human sodium/iodide-symporter gene responds to retinoic acid. Mol Cell Endocrinol 2002;189:145–155. ArticlePubMed
- 73. Grunwald F, Menzel C, Bender H, Palmedo H, Otte R, Fimmers R, et al. Redifferentiation therapy-induced radioiodine uptake in thyroid cancer. J Nucl Med 1998;39:1903–1906. PubMed
- 74. Simon D, Koehrle J, Reiners C, Boerner AR, Schmutzler C, Mainz K, et al. Redifferentiation therapy with retinoids: therapeutic option for advanced follicular and papillary thyroid carcinoma. World J Surg 1998;22:569–574. ArticlePubMedPDF
- 75. Simon D, Korber C, Krausch M, Segering J, Groth P, Gorges R, et al. Clinical impact of retinoids in redifferentiation therapy of advanced thyroid cancer: final results of a pilot study. Eur J Nucl Med Mol Imaging 2002;29:775–782. ArticlePubMedPDF
- 76. Kim WG, Kim EY, Kim TY, Ryu JS, Hong SJ, Kim WB, et al. Redifferentiation therapy with 13-cis retinoic acids in radioiodine-resistant thyroid cancer. Endocr J 2009;56:105–112. ArticlePubMed
- 77. Courbon F, Zerdoud S, Bastie D, Archambaud F, Hoff M, Eche N, et al. Defective efficacy of retinoic acid treatment in patients with metastatic thyroid carcinoma. Thyroid 2006;16:1025–1031. ArticlePubMed
- 78. Corton JC, Anderson SP, Stauber A. Central role of peroxisome proliferator-activated receptors in the actions of peroxisome proliferators. Annu Rev Pharmacol Toxicol 2000;40:491–518. ArticlePubMed
- 79. Roberts-Thomson SJ. Peroxisome proliferator-activated receptors in tumorigenesis: targets of tumour promotion and treatment. Immunol Cell Biol 2000;78:436–441. ArticlePubMed
- 80. Frohlich E, Machicao F, Wahl R. Action of thiazolidinediones on differentiation, proliferation and apoptosis of normal and transformed thyrocytes in culture. Endocr Relat Cancer 2005;12:291–303. ArticlePubMed
- 81. Park JW, Zarnegar R, Kanauchi H, Wong MG, Hyun WC, Ginzinger DG, et al. Troglitazone, the peroxisome proliferator-activated receptor-gamma agonist, induces antiproliferation and redifferentiation in human thyroid cancer cell lines. Thyroid 2005;15:222–231. ArticlePubMed
- 82. Kitazono M, Robey R, Zhan Z, Sarlis NJ, Skarulis MC, Aikou T, et al. Low concentrations of the histone deacetylase inhibitor, depsipeptide (FR901228), increase expression of the Na(+)/I(−) symporter and iodine accumulation in poorly differentiated thyroid carcinoma cells. J Clin Endocrinol Metab 2001;86:3430–3435. ArticlePubMed
- 83. Kelly WK, O'Connor OA, Krug LM, Chiao JH, Heaney M, Curley T, et al. Phase I study of an oral histone deacetylase inhibitor, suberoylanilide hydroxamic acid, in patients with advanced cancer. J Clin Oncol 2005;23:3923–3931. ArticlePubMed
- 84. Amiri-Kordestani L, Luchenko V, Peer CJ, Ghafourian K, Reynolds J, Draper D, et al. Phase I trial of a new schedule of romidepsin in patients with advanced cancers. Clin Cancer Res 2013;19:4499–4507. ArticlePubMedPMC
- 85. Nilubol N, Merkel R, Yang L, Patel D, Reynolds JC, Sadowski SM, et al. A phase II trial of valproic acid in patients with advanced, radioiodine-resistant thyroid cancers of follicular cell origin. Clin Endocrinol (Oxf) 2017;86:128–133. ArticlePubMed
- 86. Liu D, Hu S, Hou P, Jiang D, Condouris S, Xing M. Suppression of BRAF/MEK/MAP kinase pathway restores expression of iodide-metabolizing genes in thyroid cells expressing the V600E BRAF mutant. Clin Cancer Res 2007;13:1341–1349. ArticlePubMed
- 87. Hayes DN, Lucas AS, Tanvetyanon T, Krzyzanowska MK, Chung CH, Murphy BA, et al. Phase II efficacy and pharmacogenomic study of Selumetinib (AZD6244; ARRY-142886) in iodine-131 refractory papillary thyroid carcinoma with or without follicular elements. Clin Cancer Res 2012;18:2056–2065. ArticlePubMedPMC
- 88. Rothenberg SM, McFadden DG, Palmer EL, Daniels GH, Wirth LJ. Redifferentiation of iodine-refractory BRAF V600E-mutant metastatic papillary thyroid cancer with dabrafenib. Clin Cancer Res 2015;21:1028–1035. ArticlePubMed
- 89. Furuya F, Shimura H, Suzuki H, Taki K, Ohta K, Haraguchi K, et al. Histone deacetylase inhibitors restore radioiodide uptake and retention in poorly differentiated and anaplastic thyroid cancer cells by expression of the sodium/iodide symporter thyroperoxidase and thyroglobulin. Endocrinology 2004;145:2865–2875. ArticlePubMedPDF
- 90. Pugliese M, Fortunati N, Germano A, Asioli S, Marano F, Palestini N, et al. Histone deacetylase inhibition affects sodium iodide symporter expression and induces 131I cytotoxicity in anaplastic thyroid cancer cells. Thyroid 2013;23:838–846. ArticlePubMed
- 91. Fu H, Cheng L, Jin Y, Cheng L, Liu M, Chen L. MAPK inhibitors enhance HDAC inhibitor-induced redifferentiation in papillary thyroid cancer cells harboring BRAF (V600E): an in vitro study. Mol Ther Oncolytics 2019;12:235–245. ArticlePubMedPMC
- 92. Nagarajah J, Le M, Knauf JA, Ferrandino G, Montero-Conde C, Pillarsetty N, et al. Sustained ERK inhibition maximizes responses of BrafV600E thyroid cancers to radioiodine. J Clin Invest 2016;126:4119–4124. ArticlePubMedPMC
- 93. Liu YY, Zhang X, Ringel MD, Jhiang SM. Modulation of sodium iodide symporter expression and function by LY294002, Akti-1/2 and rapamycin in thyroid cells. Endocr Relat Cancer 2012;19:291–304. ArticlePubMedPMC
- 94. Plantinga TS, Heinhuis B, Gerrits D, Netea MG, Joosten LA, Hermus AR, et al. mTOR inhibition promotes TTF1-dependent redifferentiation and restores iodine uptake in thyroid carcinoma cell lines. J Clin Endocrinol Metab 2014;99:E1368–E1375. ArticlePubMedPMCPDF
- 95. Montero-Conde C, Ruiz-Llorente S, Dominguez JM, Knauf JA, Viale A, Sherman EJ, et al. Relief of feedback inhibition of HER3 transcription by RAF and MEK inhibitors attenuates their antitumor effects in BRAF-mutant thyroid carcinomas. Cancer Discov 2013;3:520–533. ArticlePubMedPMC
References
Regulation of the sodium iodide symporter (NIS) upstream enhancer (NUE) at the transcriptional level in thyroid cells. TSHR, thyroid stimulating hormone receptor; AC, adenylyl cyclase; cAMP, cyclic adenosine monophosphate; PKA, protein kinase A; CRE, cAMP-response element; CREM, CRE-modulator; Ref1, apurinic apyrimidinic endonuclease redox effector factor-1; Pax8, paired box gene-8; TGFβ, transforming growth factor β; TLR4, Toll-like receptor 4; NF-κB, p65, a member of the class II nuclear factor κ-light-chain-enhancer of activated B cells, p65; PTTG1, pituitary tumor-transforming gene-1; PBF, PTTG1-binding factor.
Known pathways involved in the regulation of sodium iodide symporter (NIS) in thyroid cancer. RTK, receptor tyrosine kinase; IGF-1, insulin-like growth factor-1; TGFβ, transforming growth factor β; PTC, papillary thyroid cancer; PI3K, phosphoinositide 3-kinase; RasGRP3, Ras guanyl releasing protein 3; PAX8, paired box gene-8; MEK, mitogen-activated extracellular signal-regulated kinase; ERK, extracellular regulated protein kinase; mTOR, mechanistic target of rapamycin; VEGFA, vascular endothelial growth factor A; MET, mesenchymal to epithelial transition factor; TSP1, thrombospondin 1; TIMP3, tissue inhibitor of metalloproteinases 3.
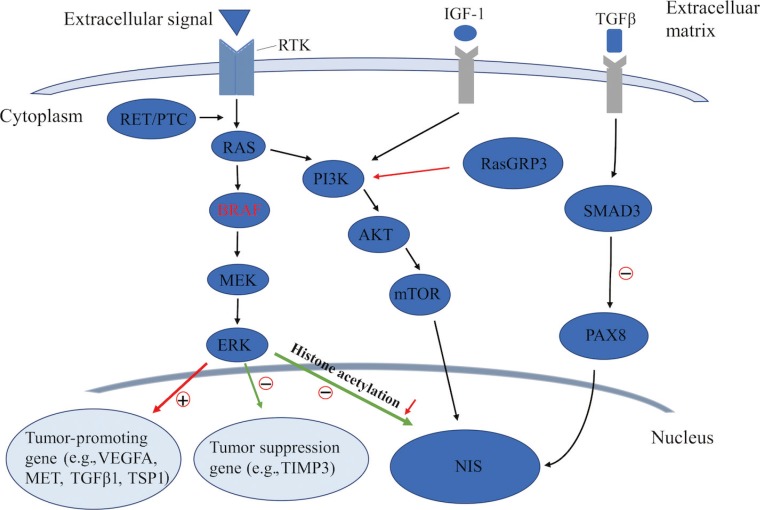
Figure & Data
References
Citations
- Long-term impact of prophylactic central neck dissection in non-invasive classic papillary thyroid carcinoma
Zehang Xu, Zhuochao Mao, Shitu Chen, Zhe Mo, Jie Zhou, Zhendong Chen, Rasa Zarnegar, Thomas J. Fahey III, Weibin Wang, Lisong Teng
European Journal of Surgical Oncology.2024; 50(1): 107305. CrossRef - The role of MAPK, notch and Wnt signaling pathways in papillary thyroid cancer: Evidence from a systematic review and meta-analyzing microarray datasets employing bioinformatics knowledge and literature
Elham Amjad, Solmaz Asnaashari, Ali Jahanban-Esfahlan, Babak Sokouti
Biochemistry and Biophysics Reports.2024; 37: 101606. CrossRef - Diagnostic Performance of [18F]TFB PET/CT Compared with Therapeutic Activity [131I]Iodine SPECT/CT and [18F]FDG PET/CT in Recurrent Differentiated Thyroid Carcinoma
David Ventura, Matthias Dittmann, Florian Büther, Michael Schäfers, Kambiz Rahbar, Daniel Hescheler, Michael Claesener, Philipp Schindler, Burkhard Riemann, Robert Seifert, Wolfgang Roll
Journal of Nuclear Medicine.2024; 65(2): 192. CrossRef - Dihydrotanshinone I exhibits antitumor effects via β-catenin downregulation in papillary thyroid cancer cell lines
Elisabetta Molteni, Federica Baldan, Giuseppe Damante, Lorenzo Allegri
Scientific Reports.2024;[Epub] CrossRef - Targeting DUSP5 suppresses malignant phenotypes of BRAF-mutant thyroid cancer cells and improves their response to sorafenib
Pu Chen, Jianling Wang, Yao Yao, Yiping Qu, Meiju Ji, Peng Hou
Endocrine.2024;[Epub] CrossRef - Lipid‐Peptide‐mRNA Nanoparticles Augment Radioiodine Uptake in Anaplastic Thyroid Cancer
Qinglin Li, Lizhuo Zhang, Jiayan Lang, Zhuo Tan, Qingqing Feng, Fei Zhu, Guangna Liu, Zhangguo Ying, Xuefei Yu, He Feng, Heqing Yi, Qingliang Wen, Tiefeng Jin, Keman Cheng, Xiao Zhao, Minghua Ge
Advanced Science.2023;[Epub] CrossRef - Case Report: Regaining radioiodine uptake following PRRT in radioiodine-refractory thyroid cancer: A new re-differentiation strategy?
Bentolhoda Hadad, Emran Askari, Seyed Rasoul Zakavi, Kamran Aryana, Soheila Erfani, Pegah Sahafi, Nima Nabavi, Atena Aghaee
Frontiers in Nuclear Medicine.2023;[Epub] CrossRef - Radioiodine therapy in advanced differentiated thyroid cancer: Resistance and overcoming strategy
Yujia Liu, Jiafeng Wang, Xiaoping Hu, Zongfu Pan, Tong Xu, Jiajie Xu, Liehao Jiang, Ping Huang, Yiwen Zhang, Minghua Ge
Drug Resistance Updates.2023; 68: 100939. CrossRef - The role of miR-139-5p in radioiodine-resistant thyroid cancer
V. Pecce, M. Sponziello, A. Verrienti, G. Grani, L. Abballe, S. Bini, S. Annunziata, G. Perotti, M. Salvatori, L. Zagaria, V. Maggisano, D. Russo, S. Filetti, C. Durante
Journal of Endocrinological Investigation.2023; 46(10): 2079. CrossRef - Anti-Cancer SERCA Inhibitors Targeting Sorafenib-Resistant Human Papillary Thyroid Carcinoma
Hang-Seok Chang, Yonjung Kim, So Young Lee, Hyeok Jun Yun, Ho-Jin Chang, Ki Cheong Park
International Journal of Molecular Sciences.2023; 24(8): 7069. CrossRef - Modulation of EZH2 Activity Induces an Antitumoral Effect and Cell Redifferentiation in Anaplastic Thyroid Cancer
Diego Claro de Mello, Kelly Cristina Saito, Marcella Maringolo Cristovão, Edna Teruko Kimura, Cesar Seigi Fuziwara
International Journal of Molecular Sciences.2023; 24(9): 7872. CrossRef - Effects of apitherapy against salivary gland disorder after radioactive iodine therapy for differentiated thyroid cancer
Kenta Nomura, Michihiro Nakayama, Atsutaka Okizaki
Annals of Nuclear Medicine.2023; 37(8): 462. CrossRef - Could Oxidative Stress Play a Role in the Development and Clinical Management of Differentiated Thyroid Cancer?
Maria Kościuszko, Angelika Buczyńska, Adam Jacek Krętowski, Anna Popławska-Kita
Cancers.2023; 15(12): 3182. CrossRef - The efficacy and safety in radioactive iodine refractory thyroid cancer patients treated with sorafenib
Yuanna Ling, Xiaoli Xiong, Jiaxin Luo, Quanliang Zou, Pan Chen, Liqin Pan, Man Long, Huijuan Feng, Wei Ouyang
Frontiers in Endocrinology.2023;[Epub] CrossRef - Advances in the molecular mechanism and targeted therapy of radioactive-iodine refractory differentiated thyroid cancer
Lu Zhang, Zhi Li, Meng Zhang, Huangren Zou, Yuke Bai, Yanlin Liu, Juan Lv, Ling Lv, Pengjie Liu, Zhiyong Deng, Chao Liu
Medical Oncology.2023;[Epub] CrossRef - Multicenter Randomized Double-Blind Phase III Trial of Donafenib in Progressive Radioactive Iodine-Refractory Differentiated Thyroid Cancer
Yansong Lin, Shukui Qin, Hui Yang, Feng Shi, Aimin Yang, Xingmin Han, Bin Liu, Zhiyong Li, Qinghai Ji, Lijun Tang, Zhiyong Deng, Yong Ding, Wei Fu, Xianhe Xie, Linfa Li, Xiaohui He, Zhongwei Lv, Qingjie Ma, Zan Shen, Zhuming Guo, Zhendong Chen, Yali Cui,
Clinical Cancer Research.2023; 29(15): 2791. CrossRef - A review of Glycogen Synthase Kinase-3 (GSK3) inhibitors for cancers therapies
Riya Thapa, Gaurav Gupta, Asif Ahmad Bhat, Waleed Hassan Almalki, Sami I. Alzarea, Imran Kazmi, Shakir Saleem, Ruqaiyah Khan, Najla Altwaijry, Harish Dureja, Sachin Kumar Singh, Kamal Dua
International Journal of Biological Macromolecules.2023; 253: 127375. CrossRef - Discovery of New Anti-Cancer Agents against Patient-Derived Sorafenib-Resistant Papillary Thyroid Cancer
Yuna Kim, Hyeok Jun Yun, Kyung Hwa Choi, Chan Wung Kim, Jae Ha Lee, Raymond Weicker, Seok-Mo Kim, Ki Cheong Park
International Journal of Molecular Sciences.2023; 24(22): 16413. CrossRef - Utilizing CD44v6 and V600EBRAF-mutation for in vitro targeted combination therapy of thyroid carcinomas
A.C.L. Mortensen, J. Imgenberg-Kreuz, D. Spiegelberg, J. Botling, M. Nestor
Heliyon.2023; 9(12): e22594. CrossRef - Canine follicular cell and medullary thyroid carcinomas: Immunohistochemical characterization
Jana Jankovic, Eve Tièche, Martina Dettwiler, Kerstin Hahn, Stephanie Scheemaeker, Martin Kessler, Sylvie Daminet, Sven Rottenberg, Miguel Campos
Veterinary Pathology.2023;[Epub] CrossRef - Advancements in theranostic applications: exploring the role of fibroblast activation protein inhibition tracers in enhancing thyroid health assessment
Yuhua Wang, Ye Liu, Huixia Geng, Wanchun Zhang
EJNMMI Research.2023;[Epub] CrossRef - Tyrosine Kinase Inhibitors for Radioactive Iodine Refractory Differentiated Thyroid Cancer
Christos Cortas, Haris Charalambous
Life.2023; 14(1): 22. CrossRef - microRNA-181a promotes the oncogene S100A2 and enhances papillary thyroid carcinoma growth by mediating the expression of histone demethylase KDM5C
Y. Wang, H. Ye, Y. Yang, J. Li, A. Cen, L. Zhao
Journal of Endocrinological Investigation.2022; 45(1): 17. CrossRef - Systemic Therapy in Thyroid Cancer
Amit Kumar Agrawal, Vanita Noronha, Vijay Patil, Nandini Menon, Akhil Kapoor, Anuradha Chougule, Pratik Chandrani, Kumar Prabhash
Indian Journal of Surgical Oncology.2022; 13(1): 68. CrossRef - Small activating RNA-activated NIS gene promotes 131I uptake and inhibits thyroid cancer via AMPK/mTOR pathway
Li Jia, Yan Chen, Fukun Chen, Juan Lv, Yanling Li, Fei Hou, Zhixian Yang, Zhiyong Deng
Pathology - Research and Practice.2022; 229: 153735. CrossRef - Effects of Anti-Cancer Drug Sensitivity-Related Genetic Differences on Therapeutic Approaches in Refractory Papillary Thyroid Cancer
Hyeok Jun Yun, Minki Kim, Sang Yong Kim, Sungsoon Fang, Yonjung Kim, Hang-Seok Chang, Ho-Jin Chang, Ki Cheong Park
International Journal of Molecular Sciences.2022; 23(2): 699. CrossRef - Comprehensive Analysis of the Prognosis and Drug Sensitivity of Differentiation-Related lncRNAs in Papillary Thyroid Cancer
Wenlong Wang, Ning Bai, Xinying Li
Cancers.2022; 14(5): 1353. CrossRef - American Head and Neck Society Endocrine Surgery Section and International Thyroid Oncology Group consensus statement on mutational testing in thyroid cancer: Defining advanced thyroid cancer and its targeted treatment
David C. Shonka, Alan Ho, Ashish V. Chintakuntlawar, Jessica L. Geiger, Jong C. Park, Nagashree Seetharamu, Sina Jasim, Amr H. Abdelhamid Ahmed, Keith C. Bible, Marcia S. Brose, Maria E. Cabanillas, Kirsten Dabekaussen, Louise Davies, Dora Dias‐Santagata,
Head & Neck.2022; 44(6): 1277. CrossRef - State of the Art in the Current Management and Future Directions of Targeted Therapy for Differentiated Thyroid Cancer
Horatiu Silaghi, Vera Lozovanu, Carmen Emanuela Georgescu, Cristina Pop, Bogdana Adriana Nasui, Adriana Florinela Cătoi, Cristina Alina Silaghi
International Journal of Molecular Sciences.2022; 23(7): 3470. CrossRef - Targeting GLI1 Transcription Factor for Restoring Iodine Avidity with Redifferentiation in Radioactive-Iodine Refractory Thyroid Cancers
Ji Min Oh, Ramya Lakshmi Rajendran, Prakash Gangadaran, Chae Moon Hong, Ju Hye Jeong, Jaetae Lee, Byeong-Cheol Ahn
Cancers.2022; 14(7): 1782. CrossRef - The Role of the Kinase Inhibitors in Thyroid Cancers
Francesca Cuomo, Claudio Giani, Gilda Cobellis
Pharmaceutics.2022; 14(5): 1040. CrossRef - In Silico Screening and Validation of PDGFRA Inhibitors Enhancing Radioiodine Sensitivity in Thyroid Cancer
Xuefei Yu, Xuhang Zhu, Lizhuo Zhang, Jiang-Jiang Qin, Chunlai Feng, Qinglin Li
Frontiers in Pharmacology.2022;[Epub] CrossRef - Clinical and prognosis value of the number of metastatic lymph nodes in patients with papillary thyroid carcinoma
Ling Zhan, Hong-fang Feng, Xi-zi Yu, Ling-rui Li, Jun-long Song, Yi Tu, Jing-ping Yuan, Chuang Chen, Sheng-rong Sun
BMC Surgery.2022;[Epub] CrossRef - Radionuclide 131I-labeled albumin-indocyanine green nanoparticles for synergistic combined radio-photothermal therapy of anaplastic thyroid cancer
Xuemei Zhang, Ziyu Yan, Zhaowei Meng, Ning Li, Qiang Jia, Yiming Shen, Yanhui Ji
Frontiers in Oncology.2022;[Epub] CrossRef - Re-evaluation of the role of autophagy in thyroid cancer treatment
Darya Kazakova, Mika Shimamura, Tomomi Kurashige, Koichiro Hamada, Yuji Nagayama
Endocrine Journal.2022; 69(7): 847. CrossRef - Radiopharmaceutical Treatments for Cancer Therapy, Radionuclides Characteristics, Applications, and Challenges
Suliman Salih, Ajnas Alkatheeri, Wijdan Alomaim, Aisyah Elliyanti
Molecules.2022; 27(16): 5231. CrossRef - Potential Therapeutic Agents against Paclitaxel—And Sorafenib-Resistant Papillary Thyroid Carcinoma
Seok-Mo Kim, Keunwan Park, Jin Hong Lim, Hyeok Jun Yun, Sang Yong Kim, Kyung Hwa Choi, Chan Wung Kim, Jae Ha Lee, Raymond Weicker, Cheol-Ho Pan, Ki Cheong Park
International Journal of Molecular Sciences.2022; 23(18): 10378. CrossRef - Research Progress of BRAF V600E Gene Mutation in Papillary Thyroid Carcinoma
延泽 刘
Advances in Clinical Medicine.2022; 12(09): 8499. CrossRef - Sinomenine Hydrochloride Promotes TSHR-Dependent Redifferentiation in Papillary Thyroid Cancer
Jing Zhang, Aomei Zhao, Xi Jia, Xinru Li, Yiqian Liang, Yan Liu, Xin Xie, Xijie Qu, Qi Wang, Yuemin Zhang, Rui Gao, Yan Yu, Aimin Yang
International Journal of Molecular Sciences.2022; 23(18): 10709. CrossRef - miR-451a suppresses papillary thyroid cancer cell proliferation and invasion and facilitates apoptosis through targeting DCBLD2 and AKT1
Jiuting Tan, Chunpu Li, Lijue Ren, Xiaohui Zhu, Fei Hua, Yuming Fu
Molecular and Cellular Probes.2022; 66: 101863. CrossRef - Anxiety and depression status prior to radioactive iodine therapy among differentiated thyroid cancer patients during the COVID‑19 pandemic
Tingting Qiao, Dingwei Gao, Junyu Tong, Yun Shen, Jiayue Ma, Zhongwei Lv, Dan Li
Supportive Care in Cancer.2022; 30(12): 10169. CrossRef - Different Expression of Thyroid-Specific Proteins in Thyroid Cancer Cells between 2-Dimensional (2D) and 3-Dimensional (3D) Culture Environment
Ji Min Oh, Prakash Gangadaran, Ramya Lakshmi Rajendran, Chae Moon Hong, Jaetae Lee, Byeong-Cheol Ahn
Cells.2022; 11(22): 3559. CrossRef - Histone acetylation modifications: A potential targets for the diagnosis and treatment of papillary thyroid cancer
Chongyang Chen, Jingfang Liu
Frontiers in Oncology.2022;[Epub] CrossRef - Effect to Therapy of Sodium-Iodine Symporter Expression by Alpha-Ray Therapeutic Agent via Sodium/Iodine Symporter
Kazuko Kaneda-Nakashima, Yoshifumi Shirakami, Tadashi Watabe, Kazuhiro Ooe, Takashi Yoshimura, Atsushi Toyoshima, Yang Wang, Hiromitsu Haba, Koichi Fukase
International Journal of Molecular Sciences.2022; 23(24): 15509. CrossRef - Reinducing Radioiodine-Sensitivity in Radioiodine-Refractory Thyroid Cancer Using Lenvatinib (RESET): Study Protocol for a Single-Center, Open Label Phase II Trial
Maaike Dotinga, Dennis Vriens, Floris H. P. van Velden, Mette K. Stam, Jan W. T. Heemskerk, Petra Dibbets-Schneider, Martin Pool, Daphne D. D. Rietbergen, Lioe-Fee de Geus-Oei, Ellen Kapiteijn
Diagnostics.2022; 12(12): 3154. CrossRef - Non-Apoptotic Programmed Cell Death in Thyroid Diseases
Feihong Ji, Xinguang Qiu
Pharmaceuticals.2022; 15(12): 1565. CrossRef - Donafenib in Progressive Locally Advanced or Metastatic Radioactive Iodine-Refractory Differentiated Thyroid Cancer: Results of a Randomized, Multicenter Phase II Trial
Yan-Song Lin, Hui Yang, Yong Ding, Yi-Zhuang Cheng, Feng Shi, Jian Tan, Zhi-Yong Deng, Zhen-Dong Chen, Rong-Fu Wang, Qing-Hai Ji, Rui Huang, Lin-Fa Li
Thyroid.2021; 31(4): 607. CrossRef - Radioactive iodine therapy may not improve disease‐specific survival in follicular variant papillary thyroid cancer without distant metastasis: A propensity score‐matched analysis
Xiaofei Wang, Xun Zheng, Jingqiang Zhu, Zhihui Li, Tao Wei
Head & Neck.2021; 43(6): 1730. CrossRef - Serum CYFRA 21.1 Level Predicts Disease Course in Thyroid Cancer with Distant Metastasis
Chaiho Jeong, Jeongmin Lee, Hyukjin Yoon, Jeonghoon Ha, Min-Hee Kim, Ja-Seong Bae, Chan-Kwon Jung, Jeong-Soo Kim, Moo-Il Kang, Dong-Jun Lim
Cancers.2021; 13(4): 811. CrossRef - Radio-Iodide Treatment: From Molecular Aspects to the Clinical View
Antonio De la Vieja, Garcilaso Riesco-Eizaguirre
Cancers.2021; 13(5): 995. CrossRef - Advances of Nanomedicine in Radiotherapy
Wei Liu, Bo Chen, Haocheng Zheng, Yun Xing, Guiyuan Chen, Peijie Zhou, Liting Qian, Yuanzeng Min
Pharmaceutics.2021; 13(11): 1757. CrossRef - Treatment Effect of Combining Lenvatinib and Vemurafenib for BRAF Mutated Anaplastic Thyroid Cancer
Chae Moon Hong, Ji Min Oh, Prakash Gangadaran, Ramya Lakshmi Rajendran, Byeong-Cheol Ahn
International Journal of Thyroidology.2021; 14(2): 127. CrossRef - TNFα-mediated activation of NF-κB downregulates sodium-iodide symporter expression in thyroid cells
Márcia Faria, Rita Domingues, Francisca Paixão, Maria João Bugalho, Paulo Matos, Ana Luísa Silva, Salvatore Papa
PLOS ONE.2020; 15(2): e0228794. CrossRef - Novel Targeted Therapies for Metastatic Thyroid Cancer—A Comprehensive Review
Mohammad Al-Jundi, Shilpa Thakur, Sriram Gubbi, Joanna Klubo-Gwiezdzinska
Cancers.2020; 12(8): 2104. CrossRef - Managing radioiodine refractory thyroid cancer: the role of dosimetry and redifferentiation on subsequent I-131 therapy
Maaike Dotinga, Dennis Vriens, Floris van Velden, Linda Heijmen, James Nagarajah, Rodney Hicks, Ellen Kapiteijn, Lioe-Fee de Geus-Oei
The Quarterly Journal of Nuclear Medicine and Molecular Imaging.2020;[Epub] CrossRef - Mediator complex subunit 16 is down-regulated in papillary thyroid cancer, leading to increased transforming growth factor-β signaling and radioiodine resistance
Hongwei Gao, Peirong Bai, Lin Xiao, Mengjia Shen, Qiuxiao Yu, Yuanyuan Lei, Wenting Huang, Xiang Lin, Xinyi Zheng, Tao Wei, Yong Jiang, Feng Ye, Hong Bu
Journal of Biological Chemistry.2020; 295(31): 10726. CrossRef - Peptide Receptor Radionuclide Therapy in Patients With Differentiated Thyroid Cancer
Dong Yun Lee, Yong-il Kim
Clinical Nuclear Medicine.2020; 45(8): 604. CrossRef - Combination of peroxisome proliferator–activated receptor gamma and retinoid X receptor agonists induces sodium/iodide symporter expression and inhibits cell growth of human thyroid cancer cells
Jui-Yu Chen, Jane-Jen Wang, Hsin-Chen Lee, Chin-Wen Chi, Chen-Hsen Lee, Yi-Chiung Hsu
Journal of the Chinese Medical Association.2020; 83(10): 923. CrossRef
 PubReader
PubReader ePub Link
ePub Link Cite
Cite