Articles
- Page Path
- HOME > Endocrinol Metab > Volume 34(4); 2019 > Article
-
Review ArticleMolecular Mechanisms of Primary Aldosteronism
-
Sergei G. Tevosian1
 , Shawna C. Fox2, Hans K. Ghayee3
, Shawna C. Fox2, Hans K. Ghayee3 -
Endocrinology and Metabolism 2019;34(4):355-366.
DOI: https://doi.org/10.3803/EnM.2019.34.4.355
Published online: December 23, 2019
1Department of Physiological Sciences, College of Veterinary Medicine, University of Florida, Gainesville, FL, USA.
2Department of Pharmacotherapy and Translational Research, College of Pharmacy, University of Florida, Gainesville, FL, USA.
3Division of Endocrinology, Department of Medicine, Malcom Randall VA Medical Center, University of Florida, Gainesville, FL, USA.
- Corresponding author: Hans K. Ghayee. Division of Endocrinology, Department of Medicine, Malcom Randall VA Medical Center, University of Florida, 1600 SW Archer Rd, Suite H-2, Gainesville, FL 32608, USA. Tel: +1-352-273-8662, Fax: +1-352-273-7441, hans.ghayee@medicine.ufl.edu
Copyright © 2019 Korean Endocrine Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
- Primary aldosteronism (PA) results from excess production of mineralocorticoid hormone aldosterone by the adrenal cortex. It is normally caused either by unilateral aldosterone-producing adenoma (APA) or by bilateral aldosterone excess as a result of bilateral adrenal hyperplasia. PA is the most common cause of secondary hypertension and associated morbidity and mortality. While most cases of PA are sporadic, an important insight into this debilitating disease has been derived through investigating the familial forms of the disease that affect only a minor fraction of PA patients. The advent of gene expression profiling has shed light on the genes and intracellular signaling pathways that may play a role in the pathogenesis of these tumors. The genetic basis for several forms of familial PA has been uncovered in recent years although the list is likely to expand. Recently, the work from several laboratories provided evidence for the involvement of mammalian target of rapamycin pathway and inflammatory cytokines in APAs; however, their mechanism of action in tumor development and pathophysiology remains to be understood.
- Hypertension is a major cardiovascular risk factor estimated to be present in approximately 70% of people over age of 60 worldwide. Elevated blood pressure (BP) correlates with increased risk of heart attack, stroke, and progression to organ failure, and currently available therapies often remain insufficient to control BP in many patients [1]. Reported demographic trends in the United States population, including its aging and the ever-increasing prevalence of obesity, indicate that hypertension is going to become an even bigger and more urgent burden in the foreseeable future. Hypertension remains a major independent prognosis risk factor for adverse health outcomes including renal and cardiovascular disease, and a leading cause of morbidity and mortality worldwide.
- The problem of inadequate BP control was magnified after the publication of the results for the recent Systolic blood PRessure INterventional Trial (SPRINT), which demonstrated that in a large hypertensive population the target BP of 120/80 mm Hg is more beneficial than 140/90 mm Hg [2]. The reported prevalence of resistant hypertension among patients who are receiving antihypertensive therapy is ranging from 9% to 18%, owing to variable diagnostic approaches and the exclusion (or non-exclusion) of patients with pseudo-resistant hypertension [3].
- Despite important advances in following the strategies aimed at adherence and optimization, truly resistant or difficult-to-control hypertension undoubtedly exists and remains a major public health problem. An important contributor to resistant hypertension is hyperaldosteronism, where excess aldosterone activates the mineralocorticoid receptor (MR). Traditionally, patients who are not surgical candidates are treated with MR antagonists (MRAs) such as spironolactone. With this approach, BP control has been achieved with patients; however, this may only be one facet of treatment. Specifically, analyzing a large, retrospective cohort, investigators assessed the use of MRAs in primary aldosteronism [4]. The primary outcome of this study was incident cardiovascular event. Among patients treated with a MRA, those patients that continued to have suppressed renin had higher incidence of cardiovascular events. Patients with unsuppressed renin showed an identical risk profile to patients with essential hypertension [4]. An important consideration underscored by this study is that, in addition to BP control, assessing hormonal changes and adjusting medications in cases of secondary hypertension as in hyperaldosteronism may be valuable in providing more effective care.
INTRODUCTION
- Hypertension is traditionally divided into primary and secondary hypertension. Primary or essential hypertension accounts for approximately 90% of clinical cases. This overwhelmingly idiopathic characterization vividly underscores how little is still known about the causes of high BP, except that it is likely to have a genetic component and often associated with physical inactivity and poor diet. Where the cause of high BP can be identified, the hypertension is known as secondary [5]. The typical causes for secondary hypertension include impaired renal or endocrine function, vascular diseases, alcohol abuse, or drug-related conditions [67]. Resistant hypertension is diagnosed in patients with primary as well as secondary hypertension. Renal impairment, older age, obesity and diabetes have all been associated with primary resistant hypertension.
- The renin-angiotensin-aldosterone system (RAAS) is a physiological pathway with a well-recognized role in BP regulation. A critical part of the pathway involves renin-stimulated production of angiotensin II (AngII) that is acting in adrenal cortical cells to cause the upregulation and the release of hormone aldosterone. Aldosterone activates MR to maintain body fluid and electrolyte homeostasis and to sustain vascular resistance under conditions of water depletion. While functional RAAS pathway is essential for maintaining homeostatic state, deregulated (increased) aldosterone production is pathogenic and contributes to the development and progression of cardiovascular and renal disease.
- There has been increased research focus and clinical attention to RAAS in an attempt to inform an adequate clinical management of HTN in the past few years [38]. Many of these studies have highlighted the importance of the MR, aldosterone and mineralocorticoid signaling in maintenance and pathological consequences of resistant hypertension [91011121314]. The MR in humans is encoded by the nuclear receptor subfamily 3, group C, member 2 (NR3C2) gene. Upon interaction with its steroid ligand, MR translocates to the nucleus and activates transcription of specific genes harboring hormone response elements in their regulatory regions. Most importantly, active MR-ligand complex in the epithelial cells leads to the expression of genes encoding proteins that regulate ionic transport (mainly the amiloride-sensitive epithelial sodium channel [ENaC], Na+/K+ pump, serum and glucocorticoid induced kinase 1 [SGK1]), culminating in sodium conservation, water retention, an excretion of potassium to maintain salt balance, and increase in BP.
- The MR can be activated by two major steroid ligands produced in the adrenal cortex, mineralocorticoids (e.g., aldosterone) and glucocorticoids (e.g., cortisol). Cortisol concentration in circulation is 100 to 1,000 fold higher than that of aldosterone, and thus aldosterone-specific activation of MR requires an environment free of cortisol. To achieve aldosterone-dependent response, the MR is protected from plasma glucocorticoid excess by corticosteroid 11-beta-dehydrogenase isozyme 2/11β-hydroxysteroid dehydrogenase 2 (11β-HSD2) enzyme that converts cortisol to inactive cortisone. Action of 11ß-HSD2 creates glucocorticoid-free environment locally and makes MR action finely attuned to aldosterone concentration [15]. The role of aldosterone-MR complex in regulating homeostasis is mostly opposite to that of atrial natriuretic peptide (ANP) secreted by the heart.
- Traditionally, aldosterone action was thought to be limited to MR residing in kidney. However, experimental as well as clinical data strongly argue against this limited role for aldosterone-dependent regulation and the importance of MR in other tissues is becoming appreciated. While mice with a global MR loss die in the neonatal period from salt wasting [16], mice with renal tubule-specific MR deletion survive [1718]. These experiments support the notion that loss of extra-renal MR contributes to the hypotension and mortality in a complete MR-deficiency. Deletion of MR in cardiomyocytes demonstrated that the receptor mediates detrimental effects in the heart under pathological conditions; however, basal heart function is not affected by MR deletion [19]. Subsequent experiments also revealed a role for MR in macrophages [20] and smooth muscle cells [2122]; in contrast, mice with targeted deletion of MR in endothelial cells were unaffected [23]. Moreover, a BP-lowering effect of MRAs can be distinguished from the effects on urinary electrolyte excretion, indicating renal-independent regulation of BP by MR in humans [24] although these data have to be interpreted with caution, given a newly discovered role for the MRA independent of MR [25]. In summary, multiple tissues contribute to MR-dependent regulation to control BP homeostasis.
MINERALOCORTICOID-DEPENDENT REGULATION OF BP
- Primary aldosteronism (PA), an increase in circulating aldosterone independent of plasma renin, is a major cause of endocrine secondary hypertension and is often associated with resistant hypertension. Primary aldosteronism was originally described by Jerome Conn in 1955 [26]. The two major types of PA are recognized, together accounting for 95% of diagnosed cases. One is a unilateral aldosterone excess that is caused by an aldosterone-producing adenoma (APA); the second is bilateral aldosterone excess as a result of bilateral adrenal hyperplasia (BAH). Rarely, a patient may have adrenal cortical carcinoma producing excess aldosterone. The prevalence reported for primary hyperaldosteronism is estimated to be 5% to 10% of all hypertensive patients; this number rises to 12% to 20% in patients with resistant hypertension [272829303132]. The mean age at diagnosis for PA is 50 years, which is often due to difficult (and thus untimely) diagnosis. Primary hyperaldosteronism affects both men and women with equal prevalence [33]. A preferred course of treatment in unilateral PA is adrenalectomy, while bilateral PA normally requires a pharmacological approach using a MRA [2834]. The two different clinical treatment modes inadvertently produced a major gap in the molecular understanding of the two PA subtypes. Surgical treatment resulted in the availability of a genetic material from a large number of APA tumor samples that were analyzed using the modern genetic approaches (e.g., next generation sequencing [NGS]). The discovery of somatic mutations in unilateral APAs provided major advances in the understanding of the basis for aldosterone overproduction [35]. In contrast, adrenal specimens from BAH patients are limited and the second PA type was less informative in advancing our knowledge of the genetic make-up of the PA and BAH-specific genetic alterations.
- Familial hyperaldosteronism
- PA is observed among patients with APA and BHA, with about only 5% of cases being familial [36]. However, as in many genetic diseases, rare familial cases provided critical insight into the genetic bases of PA. It has been suggested that currently used classification of familial hyperaldosteronism (FH) into types, FH1–FH4, should only be applied once/if the causative genetic mutation is known [37].
- CYP11B1-CYP11B2 fusion (FH1)
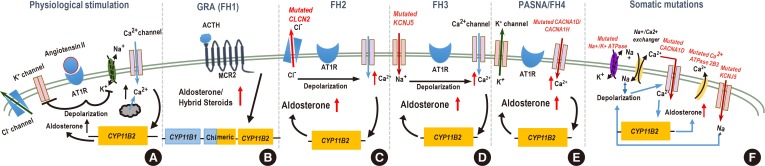
- Glucocorticoid-remediable aldosteronism (GRA) was initially described over 50 years ago [38]. Surprisingly, it was observed that aldosterone production could be suppressed by dexamethasone administration. The molecular mechanism was later elucidated in 1992 by Lifton et al. [3940]. Linkage analysis of patients' DNA revealed a fusion of highly homologous genes encoding the two key enzymes required for cortisol (11-β-hydroxylase, cytochrome P450 family 11 subfamily B member 1 [CYP11B1]) and aldosterone (aldosterone synthase, cytochrome P450 family 11 subfamily B member 2 [CYP11B2]) synthesis (Fig. 1). The recombination brings the coding regions of CYP11B2 under control of the regulatory elements of CYP11B1 that are driven by the signaling aimed at glucocorticoid synthesis (i.e., adrenocorticotropic hormone) instead of RAAS AngII and potassium. This results in aldosterone synthase expression in adrenal zona fasciculata rather than zona glomerulosa and ectopic production of aldosterone and hybrid steroids (due to oxidation by both steroid 17-alpha hydroxylase [CYP17A] and aldosterone synthase) [41].
- GRA is extremely rare and testing is recommended only for patients with an early onset of PA and in cases of familial PA occurrence or stroke at a young age (<40 years old) [2842]. The early diagnosis of GRA is important because excessive aldosterone production and associated hypertension can be corrected by low doses of dexamethasone [43]. Glucocorticoid suppression of aldosterone was previously used to both diagnose and treat GRA until introduction of the molecular techniques to detect the presence of the chimeric CYP11B1/CYP11B2 gene into the clinic [4445]. MRAs can be used as a second line of therapy to reduce deleterious non-genomic effects of aldosterone [46].
- CLCN2 (FH2)
- A different type of FH, not suppressible by glucocorticoids, has been described in several affected families with either APA or BAH [474849]. The causative genetic mutation of this type of FH remained elusive for many years. It was suggested that a locus on chromosome 7p22 was segregating with the disease, but only in some and not all cohorts. Targeted sequencing or NGS of the complete locus did not reveal any mutations [50515253]. It was the NGS analysis of the whole exome from one of the original families that uncovered a germline mutation Arg172Gln in the chloride voltage-gated channel 2 (CLCN2) gene (located on chromosome 3q27 instead) that segregated with this disease [495455]. The authors also reported other CLCN2 variants in unrelated patients. CLCN2 mutations appear to be limited to PA diagnosed at early age and it is absent in patients with essential hypertension [5455].
- CLCN2 is a member of the CLC voltage-gated Cl− channels. It is broadly expressed. CLCN2 mutations lead to leukodystrophy and infertility, and Clcn2 loss-of-function in mice leads to germ cell and photoreceptor degeneration [56575859]. Germline CLCN2 mutations that associate with PA are gain of function mutations and promote an export of Cl−, depolarization of the plasma membrane, and opening of voltage-gated Ca2+ channels, with subsequent activation of CYP11B2 transcription [5455].
- In the absence of the genetic foundation, this familial form (termed FH2) was thought to be the most prevalent, responsible for 3% to 6% of all PA cases, which, once the genetic basis was established, is considered to be a gross overestimation [60]. CLCN2 mutations were identified in about 10% of cases with young-onset PA without other known germline mutations and in 2% of patients with BAH, which suggests a significantly lower frequency than previously estimated [5455].
- KCNJ5 (FH3)
- A new inherited form of hyperaldosteronism with hyporeninemia, hypokalemia, and severe hypertension was described in several family members, a father and two young daughters, who developed hypertension by the age of 7 [61]. Adrenal glands in the patients showed notable bilateral enlargement and hypertrophy of a cortical compartment that resembled zona fasciculata juxtaposed to an atrophic zona glomerulosa. Intermixing of cells from different adrenal zones was accompanied by appearance of urine 18-oxocortisol and 18-hydroxycortisol steroids [416263]. Unlike GRA (FH1), dexamethasone administration was not effective in suppressing either glucocorticoid or mineralocorticoid synthesis. Hypertension in these patients was refractory to treatment and correction was achieved by bilateral adrenalectomy [61].
- Subsequent exome sequencing identified a heterozygous germline mutation in potassium inwardly rectifying channel subfamily J member 5 (KCNJ5) gene located on chromosome 11q24 in the original patients as well as in sporadic PA [64]. KCNJ5 encodes G-protein-activated inward rectifier K+ channel 4 (GIRK4, Kir3.4). Interaction with other Kir family members reconstitutes the functional G-protein-activated potassium channel, which controls membrane polarity in the aldosterone-producing zona glomerulosa cells. The mutation identified in the original work results in a loss of K+ selectivity and an indiscriminate influx of Na+ ions, membrane depolarization and high intracellular Ca2+ concentration. Ca2+ increase stimulates aldosterone synthesis through the activation of Ca2+ signaling pathways [656667]. The original discovery was followed by several other reports of KCNJ5 familial mutations that mostly cluster in or near protein domain responsible for channel selectivity [686970] although mutations in other locations have been documented [71727374]. The clinical characteristics of the FH3 cases vary widely, from mild to severe, in part due to the underlying mutation [7576]. Testing for FH3 is currently recommended only for patients with very early PA onset by peripheral blood DNA sequencing [28]. Treatment for FH3 varies depending on the severity of the disease, with mineralocorticoid antagonists sufficient for treating milder cases and adrenalectomy to successfully resolve resistant hypertension [7277].
- CACNA1 (FH4)
- Whole exome sequencing analysis identified a recurrent mutation in the calcium voltage-gated channel subunit alpha 1H (CACNA1H) gene in five unrelated patients in a cohort of 40 pediatric patients diagnosed with PA. That type of PA is referred to as FH4 [78] and both germline and de novo mutations of this gene have been reported [79]. Germline CACNA1H mutations have been documented in several disorders including sporadic amyotrophic lateral sclerosis, epilepsy, autism [808182]; however, for most part FH4 patients showed no symptoms that have been ascribed to these disorders [7983].
- CACNA1H encodes for the pore-forming subunit alpha 1H of the T-type calcium channel Cav3.2 that is expressed in the zona glomerulosa cells [7883] and is activated by depolarizing changes in the membrane potential [84]. The disease presentation varied in patients carrying different mutation, but most mutations are gain-of-function to some degree (although, see [85]) and augmented aldosterone production [787985]. Several patients harbored mutations in Met1549 located in the repeat domain III of Cav3.2 that is a part of a conserved methionine-phenylalanine-valine (MFV) motif that is necessary for channel inactivation [86]. Mutations in Met1549 lead to a decrease in the inactivation of the mutant Cav3.2 and a prolonged “on” state, leading to an increase in Ca2+ influx that augments the expression of CYP11B2. Treatment of HAC15 cells overexpressing Cav3.2 Met1549Val mutant channel with a T-type calcium channel blocker prevented CYP11B2 activation and aldosterone synthesis providing the basis for treatment of patients with FH4 [87]. FH4 follows an autosomal dominant pattern of inheritance, but the penetrance is limited as some Met1549 mutation carriers displayed mild or normotensive phenotype, suggesting that other factors limit the impact of gene defect [78]. Patients with CACNA1H have been also found to have developmental disorders and mental retardation [79].
- Primary Aldosteronism with Seizures and Neurologic Abnormalities (PASNA)
- Mutations have also been reported for a gene encoding a Cav1.3 subunit for another channel, CACNA1D, an L-type (high-voltage activated) Ca2+ channel expressed in zona glomerulosa [838889]. Similarly to CACNAH1, sporadic alterations in CACNA1D cause gain-of-function and an increase of Ca2+ influx with the consequent overproduction of aldosterone [8390]. In some of these patients seizures and cerebral palsy have been documented [9192]. Somatic CACNA1D was reported to be the most frequently mutated aldosterone-driver gene in African-Americans [93].
- Drivers of hyperaldosteronism
- Besides familial forms of “channelopathies” described, it is well known that somatic mutations found in aldosterone producing tumors such as KCNJ5, CACNA1, ATPase plasma membrane Ca2+ transporting 3 (ATP2B3), and ATP1A13 mutations also cause hyperaldosteronism. Mutations in KCNJ5 are considered to be the most common cause of PA overall [74] and estimated to be present in 60% to 70% cases in APA patients of Eastern Asian origin, but only about half of that in Caucasians [697394959697]. Other mutations (e.g., mutations in catenin beta 1 [CTNNB1] and protein kinase cAMP-activated catalytic subunit alpha [PRKACA] genes) lead to a loss of control over cellular proliferation and rise in the number of aldosterone-producing cells [9899100101]. Mutations in the armadillo repeat containing 5 (ARMC5) gene have been reported to cause hypercortisolism in primary bilateral macronodular adrenal hyperplasia (PBMAH) [102]. However, the connection between ARMC5 mutations and PA remains to be better understood [103]. Moreover, it remains to be determined whether somatic mutations leading to augmented aldosterone synthesis occur in an already altered adrenal cortex or they are responsible for both cellular proliferation and aldosterone production. Finally, while it is clear that BAH can be caused by the same genetic mechanisms as APA, the existence of BAH-specific pathways remains an active area of investigations. Mutations in tandem of p-domains in a weakly inward rectifying K+ channel (TWIK)-related acid-sensitive K+ channels), twik-related acid-sensitive K+ channel (TASK-1 and TASK-3) directly affect K+ balance in aldosterone-producing adrenal cells and are known to result in autonomous aldosterone production in mouse models [104105106107]; however, adrenal tumors are not formed in these animals and human disease associated with these genes have not been reported. Furthermore, evaluation of normal adrenal cortex specimens in patients without hyperaldosteronism has found that patients with essential hypertension have aldosterone producing cell clusters (APCC). Expression analysis of these APCCs revealed known aldosterone driver mutations: CACNA1D; ATP1A1, found in 35% of APCC's [108]. It is certainly worth considering if APCCs may add to the pathogenesis of hypertension.
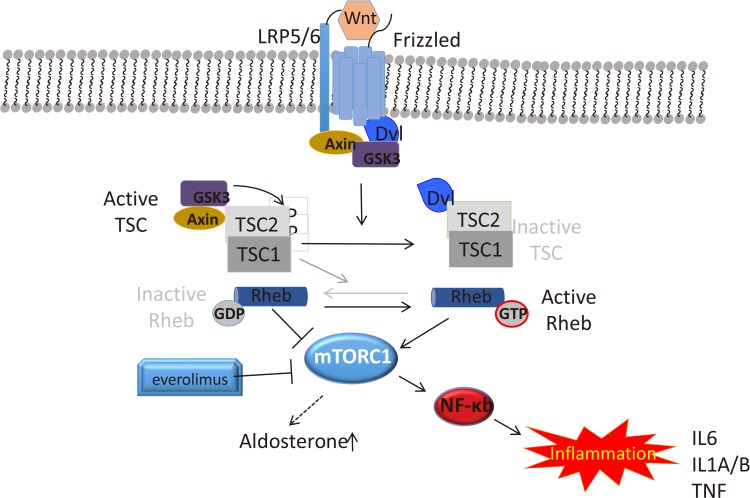
- Other area of active investigation in hyperaldosteronism explores the involvement of mammalian target of rapamycin complex 1 (mTORC1) pathway in this disease [109110]. mTORC1 is activated in a subset of patients with PA and treatment with the pathway inhibitor, everolimus, decreased BP, and increased renin concentration in patients with PA; a notable reduction of aldosterone levels was observed in some patients [111]. Similarly, tumor necrosis factor α (TNF-α) pathway is implicated in the pathogenesis of this disorder [112]. It has been long postulated that inflammatory cytokines (TNF-α and interleukins [ILs]) can play an important role in the pathogenesis of cardiovascular disease by creating an environment that promotes hypertension [113]. TNF-α and ILs act at the adrenal level in acute or chronic inflammatory states and could significantly stimulate glucocorticoid production [114115]. IL-1 receptor presence has been reported in a patient with an adrenal adenoma and tumor cells responded with cortisol hypersecretion upon IL-1ß treatment [116]. Similarly, expressions of inflammation-associated genes have been reported in APA patients [94]. It is tempting to hypothesize that mTORC and the inflammatory pathways are intertwined, at least in some tumors (Fig. 2) [117]. Our observations in adrenal cortical cell lines derived from patients' tumors confirm the importance of TNF-α signaling in adrenal neoplasia (unpublished data).
PRIMARY ALDOSTERONISM
- Hypertension is common and encountered by all clinicians in their practice. Augmented aldosterone synthesis or its amplified effect in target organs is one of the best-understood causes of secondary hypertension. Salt wasting, dehydration and hypertension are the main clinical manifestations determined mainly (but not solely) by deregulated sodium and potassium balance [118]. The long-term strategy for future research is to focus on understanding of the intrinsic and extrinsic mechanisms that result in a lack of response to BP-lowering drugs in adherent patients. As an example, the use of appropriate hormone biomarkers such as renin is helpful to determine whether patients with secondary hypertension as a result of hyperaldosteronism are effectively being treated by MR blockade. In the future, other hormones could be utilized as biomarkers to determine effective therapies and avoid early organ damage from hypertension.
- The number of gene mutations implicated in hyperaldosteronism is steadily increasing and some common mechanisms behind the PA are beginning to emerge. In addition to heritable forms of disease, APA development was found to be associated with somatic mutations. While some of these mutations are found in the same genes that are implicated in the development of the familial forms of PA (e.g., KCNJ5) others (e.g., ATP1A1 and ATP2B3) have so far only been found in the sporadic cases. Most of the mutations identified so far result in the alterations of the intracellular ionic equilibrium and deregulation of cell membrane potential. Mutations in ion pumps and channels described above, CLCN2, KCNJ5, CACNAH1, and 1D, as well as in ATP1A1, ATP2B3 are found in about 50% of APAs and figure most prominently in the PA pathogenesis [90119120121]. The functions of the affected genes provide important cues to underlying mechanisms of hypertension and will add opportunities for precision treatments in the future.
CONCLUSIONS
-
CONFLICTS OF INTEREST: No potential conflict of interest relevant to this article was reported.
Article information
- 1. Logan AG. Hypertension in aging patients. Expert Rev Cardiovasc Ther 2011;9:113–120. ArticlePubMed
- 2. SPRINT Research Group. Wright JT Jr, Williamson JD, Whelton PK, Snyder JK, Sink KM, et al. A randomized trial of intensive versus standard blood-pressure control. N Engl J Med 2015;373:2103–2116. ArticlePubMedPMC
- 3. Achelrod D, Wenzel U, Frey S. Systematic review and meta-analysis of the prevalence of resistant hypertension in treated hypertensive populations. Am J Hypertens 2015;28:355–361. ArticlePubMedPDF
- 4. Hundemer GL, Curhan GC, Yozamp N, Wang M, Vaidya A. Cardiometabolic outcomes and mortality in medically treated primary aldosteronism: a retrospective cohort study. Lancet Diabetes Endocrinol 2018;6:51–59. ArticlePubMed
- 5. Carey RM, Muntner P, Bosworth HB, Whelton PK. Prevention and control of hypertension: JACC Health Promotion Series. J Am Coll Cardiol 2018;72:1278–1293. ArticlePubMedPMC
- 6. Puar TH, Mok Y, Debajyoti R, Khoo J, How CH, Ng AK. Secondary hypertension in adults. Singapore Med J 2016;57:228–232. ArticlePubMedPMC
- 7. De Nicola L, Gabbai FB, Agarwal R, Chiodini P, Borrelli S, Bellizzi V, et al. Prevalence and prognostic role of resistant hypertension in chronic kidney disease patients. J Am Coll Cardiol 2013;61:2461–2467. ArticlePubMed
- 8. Calhoun DA, Jones D, Textor S, Goff DC, Murphy TP, Toto RD, et al. Resistant hypertension: diagnosis, evaluation, and treatment: a scientific statement from the American Heart Association Professional Education Committee of the Council for High Blood Pressure Research. Circulation 2008;117:e510–e526. PubMed
- 9. Epstein M, Calhoun DA. The role of aldosterone in resistant hypertension: implications for pathogenesis and therapy. Curr Hypertens Rep 2007;9:98–105. ArticlePubMedPDF
- 10. Sowers JR, Whaley-Connell A, Epstein M. Narrative review: the emerging clinical implications of the role of aldosterone in the metabolic syndrome and resistant hypertension. Ann Intern Med 2009;150:776–783. ArticlePubMedPMC
- 11. Duprez DA. Aldosterone and the vasculature: mechanisms mediating resistant hypertension. J Clin Hypertens (Greenwich) 2007;9:13–18. ArticlePubMedPMC
- 12. Calhoun DA. Hyperaldosteronism as a common cause of resistant hypertension. Annu Rev Med 2013;64:233–247. ArticlePubMed
- 13. Pitt B, Zannad F, Remme WJ, Cody R, Castaigne A, Perez A, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med 1999;341:709–717. ArticlePubMed
- 14. Pitt B, Remme W, Zannad F, Neaton J, Martinez F, Roniker B, et al. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med 2003;348:1309–1321. ArticlePubMed
- 15. Bailey MA. 11β-Hydroxysteroid dehydrogenases and hypertension in the metabolic syndrome. Curr Hypertens Rep 2017;19:100. ArticlePubMedPMC
- 16. Berger S, Bleich M, Schmid W, Cole TJ, Peters J, Watanabe H, et al. Mineralocorticoid receptor knockout mice: pathophysiology of Na+ metabolism. Proc Natl Acad Sci U S A 1998;95:9424–9429. ArticlePubMedPMC
- 17. Ronzaud C, Loffing J, Bleich M, Gretz N, Grone HJ, Schutz G, et al. Impairment of sodium balance in mice deficient in renal principal cell mineralocorticoid receptor. J Am Soc Nephrol 2007;18:1679–1687. ArticlePubMed
- 18. Ronzaud C, Loffing J, Gretz N, Schutz G, Berger S. Inducible renal principal cell-specific mineralocorticoid receptor gene inactivation in mice. Am J Physiol Renal Physiol 2011;300:F756–F760. ArticlePubMed
- 19. Fraccarollo D, Berger S, Galuppo P, Kneitz S, Hein L, Schutz G, et al. Deletion of cardiomyocyte mineralocorticoid receptor ameliorates adverse remodeling after myocardial infarction. Circulation 2011;123:400–408. ArticlePubMed
- 20. Lim HY, Muller N, Herold MJ, van den Brandt J, Reichardt HM. Glucocorticoids exert opposing effects on macrophage function dependent on their concentration. Immunology 2007;122:47–53. ArticlePubMedPMC
- 21. McCurley A, Pires PW, Bender SB, Aronovitz M, Zhao MJ, Metzger D, et al. Direct regulation of blood pressure by smooth muscle cell mineralocorticoid receptors. Nat Med 2012;18:1429–1433. ArticlePubMedPMCPDF
- 22. Kim SK, McCurley AT, DuPont JJ, Aronovitz M, Moss ME, Stillman IE, et al. Smooth muscle cell-mineralocorticoid receptor as a mediator of cardiovascular stiffness with aging. Hypertension 2018;71:609–621. ArticlePubMedPMC
- 23. Laursen SB, Finsen S, Marcussen N, Quaggin SE, Hansen PBL, Dimke H. Endothelial mineralocorticoid receptor ablation does not alter blood pressure, kidney function or renal vessel contractility. PLoS One 2018;13:e0193032. ArticlePubMedPMC
- 24. Funder JW, Mihailidou AS. Aldosterone and mineralocorticoid receptors: clinical studies and basic biology. Mol Cell Endocrinol 2009;301:2–6. ArticlePubMed
- 25. Good ME, Chiu YH, Poon IKH, Medina CB, Butcher JT, Mendu SK, et al. Pannexin 1 channels as an unexpected new target of the anti-hypertensive drug spironolactone. Circ Res 2018;122:606–615. ArticlePubMed
- 26. Conn JW. Presidential address. I. Painting background. II. Primary aldosteronism, a new clinical syndrome. J Lab Clin Med 1955;45:3–17. PubMed
- 27. Funder JW. Primary aldosteronism: the next five years. Horm Metab Res 2017;49:977–983. ArticlePubMedPDF
- 28. Funder JW, Carey RM, Mantero F, Murad MH, Reincke M, Shibata H, et al. The management of primary aldosteronism: case detection, diagnosis, and treatment: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 2016;101:1889–1916. ArticlePubMed
- 29. Hannemann A, Wallaschofski H. Prevalence of primary aldosteronism in patient's cohorts and in population-based studies: a review of the current literature. Horm Metab Res 2012;44:157–162. ArticlePubMedPDF
- 30. Douma S, Petidis K, Doumas M, Papaefthimiou P, Triantafyllou A, Kartali N, et al. Prevalence of primary hyperaldosteronism in resistant hypertension: a retrospective observational study. Lancet 2008;371:1921–1926. ArticlePubMed
- 31. Young WF Jr. Diagnosis and treatment of primary aldosteronism: practical clinical perspectives. J Intern Med 2019;285:126–148. ArticlePubMed
- 32. Monticone S, Burrello J, Tizzani D, Bertello C, Viola A, Buffolo F, et al. Prevalence and clinical manifestations of primary aldosteronism encountered in primary care practice. J Am Coll Cardiol 2017;69:1811–1820. ArticlePubMed
- 33. Blumenfeld JD, Sealey JE, Schlussel Y, Vaughan ED Jr, Sos TA, Atlas SA, et al. Diagnosis and treatment of primary hyperaldosteronism. Ann Intern Med 1994;121:877–885. ArticlePubMed
- 34. Katabami T, Fukuda H, Tsukiyama H, Tanaka Y, Takeda Y, Kurihara I, et al. Clinical and biochemical outcomes after adrenalectomy and medical treatment in patients with unilateral primary aldosteronism. J Hypertens 2019;37:1513–1520. ArticlePubMed
- 35. Fernandes-Rosa FL, Boulkroun S, Zennaro MC. Somatic and inherited mutations in primary aldosteronism. J Mol Endocrinol 2017;59:R47–R63. ArticlePubMed
- 36. Mulatero P, Tizzani D, Viola A, Bertello C, Monticone S, Mengozzi G, et al. Prevalence and characteristics of familial hyperaldosteronism: the PATOGEN study (Primary Aldosteronism in TOrino-GENetic forms). Hypertension 2011;58:797–803. ArticlePubMed
- 37. Perez-Rivas LG, Williams TA, Reincke M. Inherited forms of primary hyperaldosteronism: new genes, new phenotypes and proposition of a new classification. Exp Clin Endocrinol Diabetes 2019;127:93–99. ArticlePubMedPDF
- 38. Sutherland DJ, Ruse JL, Laidlaw JC. Hypertension, increased aldosterone secretion and low plasma renin activity relieved by dexamethasone. Can Med Assoc J 1966;95:1109–1119. PubMedPMC
- 39. Lifton RP, Dluhy RG, Powers M, Rich GM, Cook S, Ulick S, et al. A chimaeric 11 beta-hydroxylase/aldosterone synthase gene causes glucocorticoid-remediable aldosteronism and human hypertension. Nature 1992;355:262–265. ArticlePubMedPDF
- 40. Lifton RP, Dluhy RG, Powers M, Rich GM, Gutkin M, Fallo F, et al. Hereditary hypertension caused by chimaeric gene duplications and ectopic expression of aldosterone synthase. Nat Genet 1992;2:66–74. ArticlePubMedPDF
- 41. Lenders JWM, Williams TA, Reincke M, Gomez-Sanchez CE. Diagnosis of endocrine disease: 18-oxocortisol and 18-hydroxycortisol: is there clinical utility of these steroids? Eur J Endocrinol 2018;178:R1–R9. ArticlePubMed
- 42. Carvajal CA, Campino C, Martinez-Aguayo A, Tichauer JE, Bancalari R, Valdivia C, et al. A new presentation of the chimeric CYP11B1/CYP11B2 gene with low prevalence of primary aldosteronism and atypical gene segregation pattern. Hypertension 2012;59:85–91. ArticlePubMed
- 43. Stowasser M, Bachmann AW, Huggard PR, Rossetti TR, Gordon RD. Treatment of familial hyperaldosteronism type I: only partial suppression of adrenocorticotropin required to correct hypertension. J Clin Endocrinol Metab 2000;85:3313–3318. ArticlePubMedPDF
- 44. Litchfield WR, New MI, Coolidge C, Lifton RP, Dluhy RG. Evaluation of the dexamethasone suppression test for the diagnosis of glucocorticoid-remediable aldosteronism. J Clin Endocrinol Metab 1997;82:3570–3573. ArticlePubMed
- 45. Jonsson JR, Klemm SA, Tunny TJ, Stowasser M, Gordon RD. A new genetic test for familial hyperaldosteronism type I aids in the detection of curable hypertension. Biochem Biophys Res Commun 1995;207:565–571. ArticlePubMed
- 46. Quack I, Vonend O, Rump LC. Familial hyperaldosteronism I-III. Horm Metab Res 2010;42:424–428. ArticlePubMedPDF
- 47. Gordon RD, Stowasser M, Tunny TJ, Klemm SA, Finn WL, Krek AL. Clinical and pathological diversity of primary aldosteronism, including a new familial variety. Clin Exp Pharmacol Physiol 1991;18:283–286. ArticlePubMed
- 48. Stowasser M, Gordon RD, Tunny TJ, Klemm SA, Finn WL, Krek AL. Primary aldosteronism: implications of a new familial variety. J Hypertens Suppl 1991;9:S264–S265. PubMed
- 49. Stowasser M, Gordon RD, Tunny TJ, Klemm SA, Finn WL, Krek AL. Familial hyperaldosteronism type II: five families with a new variety of primary aldosteronism. Clin Exp Pharmacol Physiol 1992;19:319–322. ArticlePubMed
- 50. So A, Duffy DL, Gordon RD, Jeske YW, Lin-Su K, New MI, et al. Familial hyperaldosteronism type II is linked to the chromosome 7p22 region but also shows predicted heterogeneity. J Hypertens 2005;23:1477–1484. ArticlePubMed
- 51. Lafferty AR, Torpy DJ, Stowasser M, Taymans SE, Lin JP, Huggard P, et al. A novel genetic locus for low renin hypertension: familial hyperaldosteronism type II maps to chromosome 7 (7p22). J Med Genet 2000;37:831–835. ArticlePubMedPMC
- 52. Fallo F, Pilon C, Barzon L, Pistorello M, Sonino N, Veglio F, et al. Retention of heterozygosity at chromosome 7p22 and 11q13 in aldosterone-producing tumours of patients with familial hyperaldosteronism not remediable by glucocorticoids. J Hum Hypertens 2004;18:829–830. ArticlePubMedPDF
- 53. Jeske YW, So A, Kelemen L, Sukor N, Willys C, Bulmer B, et al. Examination of chromosome 7p22 candidate genes RBaK, PMS2 and GNA12 in familial hyperaldosteronism type II. Clin Exp Pharmacol Physiol 2008;35:380–385. ArticlePubMed
- 54. Scholl UI, Stolting G, Schewe J, Thiel A, Tan H, Nelson-Williams C, et al. CLCN2 chloride channel mutations in familial hyperaldosteronism type II. Nat Genet 2018;50:349–354. ArticlePubMedPMCPDF
- 55. Fernandes-Rosa FL, Daniil G, Orozco IJ, Goppner C, El Zein R, Jain V, et al. A gain-of-function mutation in the CLCN2 chloride channel gene causes primary aldosteronism. Nat Genet 2018;50:355–361. ArticlePubMedPDF
- 56. Thiemann A, Grunder S, Pusch M, Jentsch TJ. A chloride channel widely expressed in epithelial and non-epithelial cells. Nature 1992;356:57–60. ArticlePubMedPDF
- 57. Di Bella D, Pareyson D, Savoiardo M, Farina L, Ciano C, Caldarazzo S, et al. Subclinical leukodystrophy and infertility in a man with a novel homozygous CLCN2 mutation. Neurology 2014;83:1217–1218. ArticlePubMed
- 58. Depienne C, Bugiani M, Dupuits C, Galanaud D, Touitou V, Postma N, et al. Brain white matter oedema due to ClC-2 chloride channel deficiency: an observational analytical study. Lancet Neurol 2013;12:659–668. ArticlePubMed
- 59. Bosl MR, Stein V, Hubner C, Zdebik AA, Jordt SE, Mukhopadhyay AK, et al. Male germ cells and photoreceptors, both dependent on close cell-cell interactions, degenerate upon ClC-2 Cl(-) channel disruption. EMBO J 2001;20:1289–1299. ArticlePubMedPMC
- 60. Korah HE, Scholl UI. An update on familial hyperaldosteronism. Horm Metab Res 2015;47:941–946. ArticlePubMedPDF
- 61. Geller DS, Zhang J, Wisgerhof MV, Shackleton C, Kashgarian M, Lifton RP. A novel form of human mendelian hypertension featuring nonglucocorticoid-remediable aldosteronism. J Clin Endocrinol Metab 2008;93:3117–3123. ArticlePubMedPMCPDF
- 62. Gomez-Sanchez CE, Qi X, Gomez-Sanchez EP, Sasano H, Bohlen MO, Wisgerhof M. Disordered zonal and cellular CYP11B2 enzyme expression in familial hyperaldosteronism type 3. Mol Cell Endocrinol 2017;439:74–80. ArticlePubMed
- 63. Tezuka Y, Yamazaki Y, Kitada M, Morimoto R, Kudo M, Seiji K, et al. 18-Oxocortisol synthesis in aldosterone-producing adrenocortical adenoma and significance of KCNJ5 mutation status. Hypertension 2019;73:1283–1290. ArticlePubMedPMC
- 64. Choi M, Scholl UI, Yue P, Bjorklund P, Zhao B, Nelson-Williams C, et al. K+ channel mutations in adrenal aldosterone-producing adenomas and hereditary hypertension. Science 2011;331:768–772. ArticlePubMedPMC
- 65. Velarde-Miranda C, Gomez-Sanchez EP, Gomez-Sanchez CE. Regulation of aldosterone biosynthesis by the Kir3.4 (KCNJ5) potassium channel. Clin Exp Pharmacol Physiol 2013;40:895–901. ArticlePubMed
- 66. Oki K, Plonczynski MW, Luis Lam M, Gomez-Sanchez EP, Gomez-Sanchez CE. Potassium channel mutant KCNJ5 T158A expression in HAC-15 cells increases aldosterone synthesis. Endocrinology 2012;153:1774–1782. ArticlePubMedPMCPDF
- 67. Mulatero P, Monticone S, Rainey WE, Veglio F, Williams TA. Role of KCNJ5 in familial and sporadic primary aldosteronism. Nat Rev Endocrinol 2013;9:104–112. ArticlePubMedPDF
- 68. Scholl UI, Nelson-Williams C, Yue P, Grekin R, Wyatt RJ, Dillon MJ, et al. Hypertension with or without adrenal hyperplasia due to different inherited mutations in the potassium channel KCNJ5. Proc Natl Acad Sci U S A 2012;109:2533–2538. ArticlePubMedPMC
- 69. Mulatero P, Tauber P, Zennaro MC, Monticone S, Lang K, Beuschlein F, et al. KCNJ5 mutations in European families with nonglucocorticoid remediable familial hyperaldosteronism. Hypertension 2012;59:235–240. ArticlePubMed
- 70. Wang H, Weng C, Chen H. Positive association between KCNJ5 rs2604204 (A/C) polymorphism and plasma aldosterone levels, but also plasma renin and angiotensin I and II levels, in newly diagnosed hypertensive Chinese: a case-control study. J Hum Hypertens 2017;31:457–461. ArticlePubMedPDF
- 71. Murthy M, Xu S, Massimo G, Wolley M, Gordon RD, Stowasser M, et al. Role for germline mutations and a rare coding single nucleotide polymorphism within the KCNJ5 potassium channel in a large cohort of sporadic cases of primary aldosteronism. Hypertension 2014;63:783–789. ArticlePubMed
- 72. Monticone S, Bandulik S, Stindl J, Zilbermint M, Dedov I, Mulatero P, et al. A case of severe hyperaldosteronism caused by a de novo mutation affecting a critical salt bridge Kir3.4 residue. J Clin Endocrinol Metab 2015;100:E114–E118. ArticlePubMed
- 73. Zheng FF, Zhu LM, Nie AF, Li XY, Lin JR, Zhang K, et al. Clinical characteristics of somatic mutations in Chinese patients with aldosterone-producing adenoma. Hypertension 2015;65:622–628. ArticlePubMed
- 74. Williams TA, Monticone S, Mulatero P. KCNJ5 mutations are the most frequent genetic alteration in primary aldosteronism. Hypertension 2015;65:507–509. ArticlePubMed
- 75. Adachi M, Muroya K, Asakura Y, Sugiyama K, Homma K, Hasegawa T. Discordant genotype-phenotype correlation in familial hyperaldosteronism type III with KCNJ5 gene mutation: a patient report and review of the literature. Horm Res Paediatr 2014;82:138–142. ArticlePubMed
- 76. Tong A, Liu G, Wang F, Jiang J, Yan Z, Zhang D, et al. A novel phenotype of familial hyperaldosteronism type III: concurrence of aldosteronism and Cushing's syndrome. J Clin Endocrinol Metab 2016;101:4290–4297. ArticlePubMedPMC
- 77. Satoh M, Maruhashi T, Yoshida Y, Shibata H. Systematic review of the clinical outcomes of mineralocorticoid receptor antagonist treatment versus adrenalectomy in patients with primary aldosteronism. Hypertens Res 2019;42:817–824. ArticlePubMedPDF
- 78. Scholl UI, Stolting G, Nelson-Williams C, Vichot AA, Choi M, Loring E, et al. Recurrent gain of function mutation in calcium channel CACNA1H causes early-onset hypertension with primary aldosteronism. Elife 2015;4:e06315. ArticlePubMedPMC
- 79. Daniil G, Fernandes-Rosa FL, Chemin J, Blesneac I, Beltrand J, Polak M, et al. CACNA1H mutations are associated with different forms of primary aldosteronism. EBioMedicine 2016;13:225–236. ArticlePubMedPMC
- 80. Steinberg KM, Yu B, Koboldt DC, Mardis ER, Pamphlett R. Exome sequencing of case-unaffected-parents trios reveals recessive and de novo genetic variants in sporadic ALS. Sci Rep 2015;5:9124. ArticlePubMedPMCPDF
- 81. Splawski I, Yoo DS, Stotz SC, Cherry A, Clapham DE, Keating MT. CACNA1H mutations in autism spectrum disorders. J Biol Chem 2006;281:22085–22091. ArticlePubMed
- 82. Eckle VS, Shcheglovitov A, Vitko I, Dey D, Yap CC, Winckler B, et al. Mechanisms by which a CACNA1H mutation in epilepsy patients increases seizure susceptibility. J Physiol 2014;592:795–809. ArticlePubMedPMC
- 83. Scholl UI, Goh G, Stolting G, de Oliveira RC, Choi M, Overton JD, et al. Somatic and germline CACNA1D calcium channel mutations in aldosterone-producing adenomas and primary aldosteronism. Nat Genet 2013;45:1050–1054. ArticlePubMedPMCPDF
- 84. Talavera K, Nilius B. Biophysics and structure-function relationship of T-type Ca2+ channels. Cell Calcium 2006;40:97–114. ArticlePubMed
- 85. Wulczyn K, Perez-Reyes E, Nussbaum RL, Park M. Primary aldosteronism associated with a germline variant in CACNA1H. BMJ Case Rep 2019;12:e229031.ArticlePubMedPMC
- 86. Marksteiner R, Schurr P, Berjukow S, Margreiter E, Perez-Reyes E, Hering S. Inactivation determinants in segment IIIS6 of Ca(v)3.1. J Physiol 2001;537:27–34. ArticlePubMedPMC
- 87. Reimer EN, Walenda G, Seidel E, Scholl UI. CACNA1H (M1549V) mutant calcium channel causes autonomous aldosterone production in HAC15 cells and is inhibited by mibefradil. Endocrinology 2016;157:3016–3022. ArticlePubMedPDF
- 88. Xie CB, Shaikh LH, Garg S, Tanriver G, Teo AE, Zhou J, et al. Regulation of aldosterone secretion by Cav1.3. Sci Rep 2016;6:24697. ArticlePubMedPMCPDF
- 89. Lenzini L, Prisco S, Caroccia B, Rossi GP. Saga of familial hyperaldosteronism: yet a new channel. Hypertension 2018;71:1010–1014. ArticlePubMed
- 90. Azizan EA, Poulsen H, Tuluc P, Zhou J, Clausen MV, Lieb A, et al. Somatic mutations in ATP1A1 and CACNA1D underlie a common subtype of adrenal hypertension. Nat Genet 2013;45:1055–1060. ArticlePubMedPDF
- 91. Pinggera A, Mackenroth L, Rump A, Schallner J, Beleggia F, Wollnik B, et al. New gain-of-function mutation shows CACNA1D as recurrently mutated gene in autism spectrum disorders and epilepsy. Hum Mol Genet 2017;26:2923–2932. ArticlePubMedPMCPDF
- 92. Pinggera A, Lieb A, Benedetti B, Lampert M, Monteleone S, Liedl KR, et al. CACNA1D de novo mutations in autism spectrum disorders activate Cav1.3 L-type calcium channels. Biol Psychiatry 2015;77:816–822. ArticlePubMedPMC
- 93. Nanba K, Omata K, Gomez-Sanchez CE, Stratakis CA, Demidowich AP, Suzuki M, et al. Genetic characteristics of aldosterone-producing adenomas in blacks. Hypertension 2019;73:885–892. ArticlePubMedPMC
- 94. Murakami M, Yoshimoto T, Nakano Y, Tsuchiya K, Minami I, Bouchi R, et al. Expression of inflammation-related genes in aldosterone-producing adenomas with KCNJ5 mutation. Biochem Biophys Res Commun 2016;476:614–619. ArticlePubMed
- 95. Wang B, Li X, Zhang X, Ma X, Chen L, Zhang Y, et al. Prevalence and characterization of somatic mutations in Chinese aldosterone-producing adenoma patients. Medicine (Baltimore) 2015;94:e708. ArticlePubMedPMC
- 96. Taguchi R, Yamada M, Nakajima Y, Satoh T, Hashimoto K, Shibusawa N, et al. Expression and mutations of KCNJ5 mRNA in Japanese patients with aldosterone-producing adenomas. J Clin Endocrinol Metab 2012;97:1311–1319. ArticlePubMed
- 97. Wu VC, Wang SM, Chueh SJ, Yang SY, Huang KH, Lin YH, et al. The prevalence of CTNNB1 mutations in primary aldosteronism and consequences for clinical outcomes. Sci Rep 2017;7:39121. ArticlePubMedPMCPDF
- 98. Boulkroun S, Samson-Couterie B, Golib-Dzib JF, Amar L, Plouin PF, Sibony M, et al. Aldosterone-producing adenoma formation in the adrenal cortex involves expression of stem/progenitor cell markers. Endocrinology 2011;152:4753–4763. ArticlePubMedPDF
- 99. Berthon A, Stratakis CA. From β-catenin to ARM-repeat proteins in adrenocortical disorders. Horm Metab Res 2014;46:889–896. ArticlePubMedPDF
- 100. Tissier F, Cavard C, Groussin L, Perlemoine K, Fumey G, Hagnere AM, et al. Mutations of beta-catenin in adrenocortical tumors: activation of the Wnt signaling pathway is a frequent event in both benign and malignant adrenocortical tumors. Cancer Res 2005;65:7622–7627. ArticlePubMed
- 101. Akerstrom T, Maharjan R, Sven Willenberg H, Cupisti K, Ip J, Moser A, et al. Activating mutations in CTNNB1 in aldosterone producing adenomas. Sci Rep 2016;6:19546. ArticlePubMedPMC
- 102. Assie G, Libe R, Espiard S, Rizk-Rabin M, Guimier A, Luscap W, et al. ARMC5 mutations in macronodular adrenal hyperplasia with Cushing's syndrome. N Engl J Med 2013;369:2105–2114. ArticlePubMedPMC
- 103. Mulatero P, Schiavi F, Williams TA, Monticone S, Barbon G, Opocher G, et al. ARMC5 mutation analysis in patients with primary aldosteronism and bilateral adrenal lesions. J Hum Hypertens 2016;30:374–378. ArticlePubMedPDF
- 104. Davies LA, Hu C, Guagliardo NA, Sen N, Chen X, Talley EM, et al. TASK channel deletion in mice causes primary hyperaldosteronism. Proc Natl Acad Sci U S A 2008;105:2203–2208. ArticlePubMedPMC
- 105. Heitzmann D, Derand R, Jungbauer S, Bandulik S, Sterner C, Schweda F, et al. Invalidation of TASK1 potassium channels disrupts adrenal gland zonation and mineralocorticoid homeostasis. EMBO J 2008;27:179–187. ArticlePubMed
- 106. Guagliardo NA, Yao J, Hu C, Schertz EM, Tyson DA, Carey RM, et al. TASK-3 channel deletion in mice recapitulates low-renin essential hypertension. Hypertension 2012;59:999–1005. ArticlePubMedPMC
- 107. Penton D, Bandulik S, Schweda F, Haubs S, Tauber P, Reichold M, et al. Task3 potassium channel gene invalidation causes low renin and salt-sensitive arterial hypertension. Endocrinology 2012;153:4740–4748. ArticlePubMedPDF
- 108. Nishimoto K, Tomlins SA, Kuick R, Cani AK, Giordano TJ, Hovelson DH, et al. Aldosterone-stimulating somatic gene mutations are common in normal adrenal glands. Proc Natl Acad Sci U S A 2015;112:E4591–E4599. ArticlePubMedPMC
- 109. Su H, Gu Y, Li F, Wang Q, Huang B, Jin X, et al. The PI3K/AKT/mTOR signaling pathway is overactivated in primary aldosteronism. PLoS One 2013;8:e62399. ArticlePubMedPMC
- 110. Swierczynska MM, Betz MJ, Colombi M, Dazert E, Jeno P, Moes S, et al. Proteomic landscape of aldosterone-producing adenoma. Hypertension 2019;73:469–480. ArticlePubMed
- 111. Trinh B, Hepprich M, Betz MJ, Burkard T, Cavelti-Weder C, Seelig E, et al. Treatment of primary aldosteronism with mTORC1 inhibitors. J Clin Endocrinol Metab 2019;104:4703–4714. ArticlePubMedPDF
- 112. Hantel C, Ozimek A, Lira R, Ragazzon B, Jackel C, Frantsev R, et al. TNF alpha signaling is associated with therapeutic responsiveness to vascular disrupting agents in endocrine tumors. Mol Cell Endocrinol 2016;423:87–95. ArticlePubMed
- 113. Harrison DG, Guzik TJ, Lob HE, Madhur MS, Marvar PJ, Thabet SR, et al. Inflammation, immunity, and hypertension. Hypertension 2011;57:132–140. ArticlePubMed
- 114. Path G, Bornstein SR, Ehrhart-Bornstein M, Scherbaum WA. Interleukin-6 and the interleukin-6 receptor in the human adrenal gland: expression and effects on steroidogenesis. J Clin Endocrinol Metab 1997;82:2343–2349. ArticlePubMed
- 115. Mikhaylova IV, Kuulasmaa T, Jaaskelainen J, Voutilainen R. Tumor necrosis factor-alpha regulates steroidogenesis, apoptosis, and cell viability in the human adrenocortical cell line NCI-H295R. Endocrinology 2007;148:386–392. ArticlePubMedPDF
- 116. Willenberg HS, Stratakis CA, Marx C, Ehrhart-Bornstein M, Chrousos GP, Bornstein SR. Aberrant interleukin-1 receptors in a cortisol-secreting adrenal adenoma causing Cushing's syndrome. N Engl J Med 1998;339:27–31. ArticlePubMed
- 117. Sethi JK, Vidal-Puig A. Wnt signalling and the control of cellular metabolism. Biochem J 2010;427:1–17. ArticlePubMedPMCPDF
- 118. Bonvalet JP. Regulation of sodium transport by steroid hormones. Kidney Int Suppl 1998;65:S49–S56. PubMed
- 119. Beuschlein F, Boulkroun S, Osswald A, Wieland T, Nielsen HN, Lichtenauer UD, et al. Somatic mutations in ATP1A1 and ATP2B3 lead to aldosterone-producing adenomas and secondary hypertension. Nat Genet 2013;45:440–444.e1-2. ArticlePubMedPDF
- 120. Prada ETA, Burrello J, Reincke M, Williams TA. Old and new concepts in the molecular pathogenesis of primary aldosteronism. Hypertension 2017;70:875–881. ArticlePubMed
- 121. Zennaro MC, Boulkroun S, Fernandes-Rosa F. An update on novel mechanisms of primary aldosteronism. J Endocrinol 2015;224:R63–R77. ArticlePubMed
References
(A) Normally, AT1 receptor (AT1R) activation induces depolarization as a result of inactivation of the potassium channel Kir3.4 and Na+K+-ATPase. This depolarization triggers influx of Ca2+ through voltage-gated Ca2+ channels (e.g., calcium channel, voltage-dependent, L type, alpha 1D subunit [Cav1.3]), and the resultant rise in intracellular Ca2+ activates the aldosterone synthase gene cytochrome P450 family 11 subfamily B member 2 (CYP11B2). Ca2+-ATPase (e.g., ATPase plasma membrane Ca2+ transporting 3 [ATP2B3]) subsequently shuttles Ca2+ outside the cell. (B) In cells harboring CYP11B1-CYP11B2 gene fusion event, increased CYP11B2 synthesis is controlled by the adrenocorticotropic hormone (ACTH) through its melanocortin receptor 2 (MCR2) receptor. (C) Germline gain of function mutations in chloride voltage-gated channel 2 (CLCN2) chloride channel promotes export of Cl− ions, depolarization of the plasma membrane, and opening of voltage-gated Ca2+ channels. (D) Potassium inwardly rectifying channel subfamily J member 5 (KCNJ5) mutations change the selectivity of the Kir3.4 allowing for Na+ influx. Increased concentration of Na+ ions causes depolarization in the absence of AT1R stimulation. (E) Mutations in calcium voltage-gated channel subunit alpha1 D/H (CACNA1D/1H) subunits increase conductance of Ca2+, (F) whereas mutations in ATP2B3 prevent removal of Ca2+ ions from the cell; both of these mutations increase intracellular Ca2+ concentration and activate CYP11B2 transcription and aldosterone synthesis. Ion channel mutations (encoded by the CLCN2, KCNJ5, CACNA1D/1H, and ATP2B3) in the adrenal glomerulosa cell are linked to excessive aldosterone production. FH2, familial hyperaldosteronism type 2; PASNA, Primary Aldosteronism with Seizures and Neurologic Abnormalities.

Wnt-induced activation of Frizzled (Fzd) leads to inhibition of glycogen synthase kinase 3 (GSK3) in a ß-catenin independent way. GSK3 inhibition inactivates its substrate TSC, TSC2 (tuberous sclerosis complex 2 or tuberin). TSC2 is a tumor suppressor complex that controls mammalian target of rapamycin (mTOR) activity through the regulatory Rheb protein. In addition to Fzd, LDL receptor related protein 5/6 (LRP5/6) co-receptor and GSK3, the scaffold proteins of the canonical Wnt cascade, Dvl, and Axin, have been implicated in signal transduction. mTOR activation can in turn cause upregulation of the inflammatory pathways through nuclear factor κb (NF-κb) and increased aldosterone synthesis through yet unknown mechanism. GDP, guanosine-5′-triphosphate; mTORC1, mammalian target of rapamycin complex 1; GTP, guanosine-5′-diphosphate; IL, interleukin; TNF, tumor necrosis factor.

Figure & Data
References
Citations
- Mineralocorticoid Receptor and Aldosterone: Interaction Between NR3C2 Genetic Variants, Sex, and Age in a Mixed Cohort
Mahyar Heydarpour, Wasita W Parksook, Luminita H Pojoga, Gordon H Williams, Jonathan S Williams
The Journal of Clinical Endocrinology & Metabolism.2024;[Epub] CrossRef - Proteomic analysis of urinary extracellular vesicles highlights specific signatures for patients with primary aldosteronism
Lorenzo Bertolone, Annalisa Castagna, Marcello Manfredi, Domenica De Santis, Francesca Ambrosani, Elisa Antinori, Paolo Mulatero, Elisa Danese, Emilio Marengo, Elettra Barberis, Mariangela Veneri, Nicola Martinelli, Simonetta Friso, Francesca Pizzolo, Oli
Frontiers in Endocrinology.2023;[Epub] CrossRef - Focus on adrenal and related causes of hypertension in childhood and adolescence: Rare or rarely recognized?
Flávia A. Costa-Barbosa, Rafael B. Giorgi, Claudio E. Kater
Archives of Endocrinology and Metabolism.2022;[Epub] CrossRef - Diseases caused by mutations in the Na+/K+ pump α1 gene ATP1A1
Elisa D. Biondo, Kerri Spontarelli, Giovanna Ababioh, Lois Méndez, Pablo Artigas
American Journal of Physiology-Cell Physiology.2021; 321(2): C394. CrossRef - Aldosterone Inhibits In Vitro Myogenesis by Increasing Intracellular Oxidative Stress via Mineralocorticoid Receptor
Jin Young Lee, Da Ae Kim, Eunah Choi, Yun Sun Lee, So Jeong Park, Beom-Jun Kim
Endocrinology and Metabolism.2021; 36(4): 865. CrossRef - Progress on Genetic Basis of Primary Aldosteronism
Izabela Karwacka, Łukasz Obołończyk, Sonia Kaniuka-Jakubowska, Michał Bohdan, Krzysztof Sworczak
Biomedicines.2021; 9(11): 1708. CrossRef
 PubReader
PubReader ePub Link
ePub Link Cite
Cite