Implications of Mitochondrial Unfolded Protein Response and Mitokines: A Perspective on Fatty Liver Diseases
Article information

Abstract
The signaling network of the mitochondrial unfolded protein response (UPRmt) and mitohormesis is a retrograde signaling pathway through which mitochondria-to-nucleus communication occurs in organisms. Recently, it has been shown that the UPRmt is closely associated with metabolic disorders and conditions involving insulin resistance, such as alcoholic and non-alcoholic fatty liver and fibrotic liver disease. Scientific efforts to understand the UPRmt and mitohormesis, as well as to establish the mitochondrial proteome, have established the importance of mitochondrial quality control in the development and progression of metabolic liver diseases, including non-alcoholic fatty liver disease (NAFLD) and non-alcoholic steatohepatitis (NASH). In this review, we integrate and discuss the recent data from the literature on the UPRmt and mitohormesis in metabolic liver diseases, including NAFLD/NASH and fibrosis.
INTRODUCTION
Metabolic liver diseases constitute a major health burden. Among them, fatty liver, characterized by the presence of large lipid vacuoles within the cytosol in hepatocytes, is a benign condition, but pathognomonic for non-alcoholic fatty liver disease (NAFLD) and alcoholic liver injury. Despite the globally widespread incidence and prevalence of NAFLD and non-alcoholic steatohepatitis (NASH), almost no pharmacotherapeutic options are available for these conditions. In approximately 10% of patients, NAFLD can progress in the long term to NASH, which can lead to fibrosis, cirrhosis, and hepatocellular carcinoma [1].
Liver parenchymal cells display a prominent eosinophilic cytoplasm upon hematoxylin-eosin staining, indicating that they have abundant mitochondria [2]. This means that mitochondrial dysfunction in hepatocytes results in cellular damage, which is linked to fatty liver diseases. For instance, chronic alcohol consumption alters mitochondrial oxidative phosphorylation in the liver by suppressing the synthesis of respiratory complex proteins [3], and hepatic mitochondria are structurally and functionally altered in NAFLD [4]. Thus, it is not surprising that mitochondrial dysfunction involving structural and molecular alterations leads to metabolic disturbances, potentially resulting in metabolic liver diseases, ranging from fatty liver to hepatocellular carcinoma.
Recently, one of the most remarkable scientific discoveries in mitochondrial biology has been the identification of the mitochondrial unfolded protein response (UPRmt), a retrograde mitochondria-to-nucleus signaling pathway induced by mitochondrial proteotoxic stress (Fig. 1). The UPRmt was first identified in the late 1990s, and is an area of increased scientific interest for research on aging-related degenerative diseases and metabolic disorders in organisms ranging from worms to mammals [5]. Regarding liver diseases, eliciting the UPRmt or mitohormetic factors not only inhibits the progression of NAFLD [6], but also ameliorates hepatic fibrosis and alcoholic liver injury in mice [7].
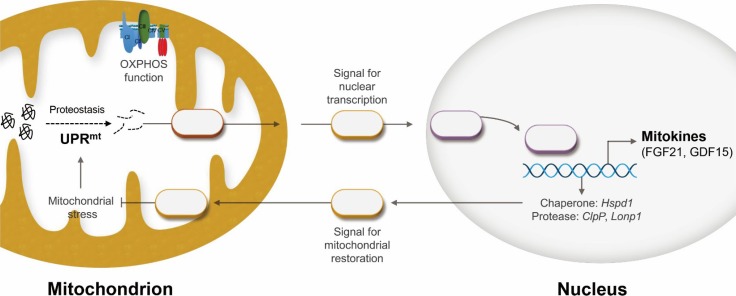
Scheme depicting the mitochondrial unfolded protein response (UPRmt) and mitokines. As a cell autonomous response, damaged mitochondria within a cell communicate to the nucleus via retrograde signaling. In contrast, mitokines (as a cell non-autonomous factor) are induced during the UPRmt and are secreted from cells affected by mitochondrial stress. OXPHOS, oxidative phosphorylation; FGF21, fibroblast growth factor 21; GDF15, growth differentiation factor 15; Hspd1, heat shock protein family D member 1; ClpP, caseinolytic peptidase P; Lonp1, LON protease 1.
In this review, we summarize the current knowledge on the implications of mitochondrial function and protein homeostasis in the development of metabolic liver diseases, including NAFLD/NASH, alcoholic liver disease, and fibrosis. We also discuss mitochondrial proteostasis and mitohormesis as new therapeutic targets for the treatment of a broad spectrum of metabolic liver diseases.
MITOCHONDRIAL BIOLOGY IN FATTY LIVER AND FIBROTIC LIVER DISEASES
Metabolic liver diseases are associated with functional alterations of mitochondrial oxidative phosphorylation [89]. For example, hepatic depletion of mitochondrial flavoprotein apoptosis inducing factor induced deficient oxidative phosphorylation, but resulted in the improvement of hepatic insulin resistance in mice [10]. In contrast, alcohol-mediated mitochondrial DNA (mtDNA) damage deteriorated cellular energy metabolism via enhanced formation of reactive oxygen species (ROS) in mice [11]. Moreover, in another mouse study, carbon tetrachloride (CCl4) decreased mitochondrial respiratory chain complex IV activity and reduced hepatic mtDNA [12]. Furthermore, the ROS produced by cytochrome P450 2E1-mediated CCl4 metabolism has been found to bind to mtDNA and promote lipid peroxidation, leading to mtDNA degradation [12]. Thus, ROS-mediated hepatocellular oxidative damage is implicated in the progression of NAFLD and fibrotic liver diseases.
RECENT INSIGHTS INTO MITOCHONDRIAL PROTEOSTASIS AND MITOKINES
Recent investigations in worms and mammals have demonstrated that genetic inhibition of mitochondrial oxidative phosphorylation activated the UPRmt, leading to an increased lifespan and improved metabolic phenotypes [13]. This suggests that modest mitochondrial inhibition paradoxically improves the metabolic phenotype and lifespan across species. This phenomenon, which is defined as mitochondrial homeostasis, or mitohormesis, involves the UPRmt and plays a prominent role in aging and degenerative diseases. Many scientists are trying to establish the role of the UPRmt in mammalian cells and systems, and to characterize its possible role in mitohormesis [5]. The mitochondrial chaperones and proteases predicted to be involved in the regulation of mitochondrial protein homeostasis may play a pivotal role in the modulation of fatty liver diseases in mammalian systems [5]. While elucidation of the UPRmt has contributed to a greater understanding of the role of mitochondria in stress adaptation and lifespan regulation, important questions remain unanswered about the role of the UPRmt in the development of fatty liver and fibrotic liver diseases.
ASPECTS OF THE CELL AUTONOMOUS RESPONSE
Mitochondrial chaperones
Mitochondrial chaperones and proteases play important roles in normal cellular function and survival, which are required for maintaining organismal homeostasis. Emerging studies have shown that the biological effects of UPRmt activation in conditions of increased mitochondrial proteotoxic stress are mediated by both cell autonomous and cell non-autonomous factors [14]. In the liver, heat shock protein 60 (HSP60) is involved in NAFLD/NASH, chronic hepatitis, and liver cancer [15]. Hsp60 works as a major defense system against proteotoxic cellular damage during alcoholic liver injury [16]. Heat shock protein family D member 1 (Hspd1) was also found to be an essential factor in regulating the inflammatory response and cell proliferation during tissue regeneration in zebrafish [17]. Deficiency of Hspd1 resulted in acute hepatic injury and cholangiocellular tumorigenesis via hepatic mitochondrial dysfunction [18]. In contrast, Hsp90 increased lipid accumulation in the liver by modulating peroxisome proliferator-activated receptor γ (PPARγ) activity in a mouse model of NAFLD [19]. Additionally, a recent proteomic analysis found that both Hsp90 isoforms were elevated in patients with NAFLD, suggesting a link between NAFLD and Hsp90 in humans [20]. Deletion of prohibitin 1 in the liver promotes liver injury, oxidative stress, inflammation, and fibrosis with development of liver cancer [2122]. HSP72 is also involved in chronic liver diseases. HSP72 mRNA expression in skeletal muscle was found to be negatively correlated with the insulin-stimulated glucose disposal rate in patients with type 2 diabetes in a hyperinsulinemic-euglycemic clamp study [23]. Patients with NASH or chronic hepatitis C virus infection showed an increase in HSP72 levels [24]. Mice with hepatocyte-specific Hsp72 overexpression were protected from acute or chronic liver injury induced by a single injection of acetaminophen, a methionine-/choline-deficient diet, or a 3,5-diethoxycarbonyl-1,4-dihydrocollidine-supplemented diet [24].
Mitochondrial proteases
In addition to the mitochondrial chaperone system, many kinds of proteases are also evolutionarily conserved for mitochondrial quality control. Similar to chaperones, mitochondrial proteases are predicted to be involved in the regulation of mitochondrial protein homeostasis, thereby playing an essential role in the modulation of metabolic diseases across species. The inner mitochondrial membrane protease OMA1 zinc metallopeptidase (OMA1) plays an essential role in the proteolytic inactivation of the GTPase optic atrophy protein 1 (OPA1), which regulates mitochondrial quality control. A remarkable level of steatosis was found in the liver of Oma1-deficient mice on a normal chow diet, and was further significantly elevated under a high-fat diet [25]. OPA1 is required for fusion of the outer mitochondrial membrane and inner mitochondrial membrane in mammals [26]. Acute Opa1 deletion in skeletal muscle was found to induce hepatic steatosis and high levels of inflammatory cytokines in mice [27]. Mitofusin-2-mediated proteolytic inactivation of Opa1 was found to be associated with mitochondria-endoplasmic reticulum contact assembly in the postprandial liver [28]. LON protease (also known as LONP1) is an ATP-dependent serine protease that is essential for mitochondrial protein homeostasis within the mitochondrial matrix. Reduction of LONP1 by small interfering RNA increased intracellular ROS production in liver cells [29]. Downregulation of mitochondrial LON protease also promoted hepatic glucose production by inducing glucose-6-phosphatase and peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) in human liver SK-HEP-1 cells [29]. Moreover, deficiency of LONP1 also resulted in gluconeogenesis by induction of glucose-6-phosphatase and PGC-1α in human liver SK-HEP-1 cells [29]. Furthermore, ATP-stimulated LONP1 activity was remarkably reduced in the hepatic mitochondrial matrix with aging in rats [30]. Caseinolytic peptidase P (ClpP), another matrix protease, plays a crucial role in the initiation of the mammalian UPRmt. ClpP−/− mice showed reduced adiposity and enhanced insulin-stimulated Akt phosphorylation in the liver [31]. ClpP−/− mice were protected against diet-induced obesity, insulin resistance, and fatty liver under a high-fat diet [31]. This paradoxical beneficial effect of ClpP deficiency is due to mitochondrial matrix proteostasis through upregulation of mitochondrial chaperones and proteases.
ASPECTS OF MITOHORMESIS: MITOKINES
Fibroblast growth factor 21
Remarkable studies in worms have established a class of secretory proteins defined as mitochondrial cytokines or mitokines [32]. Mitokine responses in mammalian system can be regarded as critical cell non-autonomous factors in disease development and progression. Individual mitokines can be used as disease markers, and these mitohormetic factors may serve as potential therapeutics for metabolic diseases including NAFLD/NASH and insulin resistance [5].
Fibroblast growth factor 21 (FGF21) was the first mitokine reported in mammalian systems. Under physiologic perturbations such as fasting, the liver is the most important organ that secretes FGF21 into the circulation, suggesting that circulating FGF21 derives largely from the liver [33]. However, FGF21 expression is markedly induced in muscle myopathies induced by mitochondrial stress [7]. Specifically, FGF21 stimulates β-oxidation of fatty acids, as well as the production of hepatic ketone bodies, and suppresses lipogenesis in the liver [34].
Impaired mitochondrial function resulted in activating transcription factor 4 (ATF4)-dependent FGF21 induction, leading to protection from diet-induced obesity and hepatic steatosis in mice [35]. In addition, FGF21 inhibited hepatic steatosis and enhanced hepatic insulin sensitivity by lowering hepatic gluconeogenesis and increasing hepatic glycogen content, thereby leading to improvements in systemic glucose intolerance and insulin resistance in mice with diet-induced obesity [3637]. Moreover, FGF21 reduced low density lipoprotein cholesterol and increased high density lipoprotein cholesterol in the serum of non-human primates [38]. FGF21 has been found to ameliorate alcohol-induced fatty liver by activating the AMP-activated protein kinase-sirtuin 1 pathway, and by attenuating intracellular ROS production [39]. Furthermore, administration of recombinant human FGF21 decreased chronic alcohol-induced hepatic steatosis and inflammation through increased fatty acid β-oxidation and decreased lipogenesis in mice [40]. FGF21-deficient mice were found to have worsened steatosis, inflammation, and fibrosis under a methionine- and choline-deficient diet for 8 to 16 weeks [41]. Treating mice with recombinant FGF21 also decreased hepatic fibrosis and prevented induction of fibrotic mediators in the liver by regulating the transforming growth factor β (TGF-β)/Smad2/3 and nuclear factor κB (NF-κB) signaling pathways [42].
The receptors for FGF21 consists of FGF receptor (FGFR) and βKlotho, which activate FGF21 signal transduction [43]. Among FGFRs widely distributed in the body, FGFR1c is a key receptor for FGF21, and other receptors mediating FGF21 signaling include FGFR2 and FGFR4 [44]. Tissue-specific expression of βKlotho may determine the biological functions of FGF21 because FGFRs are ubiquitously expressed in the many kinds of tissues [45].
Growth differentiation factor 15
Growth differentiation factor 15 (GDF15), a member of the TGF-β superfamily, has anti-inflammatory activities in mammalian systems [46]. Recently, dysfunctional mitochondrial oxidative phosphorylation has been found to promote the induction of GDF15, a mitokine regulating systemic energy metabolism in mice [47]. C/EBP-homologous protein (CHOP), which is part of the unfolded protein response signaling pathway in the mitochondria and endoplasmic reticulum, directly binds to the promoter of GDF15 and activates its transcription [4748]. GDF15 is induced by the cellular adaptive response to tissue injury or exposure to carcinogens in a number of solid organs [49]. Additionally, GDF15 as a serum biomarker predicts liver diseases including NAFLD, NASH, and advanced liver fibrosis in humans [50]. Elevated levels of GDF15 are also closely associated with increased risk of chronic liver and cardiovascular diseases [5].
Treatment with recombinant GDF15 reduced fat accumulation in the liver and attenuated NAFLD progression by enhancing hepatic fatty acid β-oxidation in mice fed a high-fat diet [4751]. In contrast, abrogation of hepatic Gdf15 decreased fatty acid β-oxidation and ketone production in fasting mice [52]. Knockdown of hepatic Gdf15 also reduced ketogenesis in streptozotocin-induced diabetic mice, as well as mice fed a ketogenic diet [52]. GDF15 deficiency exacerbated hepatic fat deposition and the induction of pro-inflammatory cytokines in mice fed a liquid ethanol diet [7]. Moreover, GDF15 deficiency promoted liver inflammation and the expression of fibrotic mediators in a CCl4-induced mouse model [7]. Furthermore, GDF15 inhibited the growth and activity of fibroblasts via the modulating TGF-β-Smad signaling pathway in fibroblasts [53].
Recently, several emerging studies have identified that GDNF family receptor α-like (GFRAL) in the hindbrain is the receptor for GDF15 and a therapeutic target for the treatment of obesity and metabolic diseases [545556]. However, the receptors for GDF15 in peripheral tissues have not yet been identified, although GDF15 has been shown to play a variety of roles in peripheral organs. Thus, further studies are required to understand the peripheral roles of GDF15 in metabolic organs such as the liver, adipose tissue, and skeletal muscle.
UPRmt AND MITOHORMESIS AS NEW THERAPEUTIC TARGETS FOR FATTY LIVER DISEASES
Modest or transient mitochondrial stress induces UPRmt and mitohormesis in mammalian and worms. This response may provide protection or amelioration to the host in both a cell autonomous and cell non-autonomous manner. Thus, identification of UPRmt regulators may serve to restore or prevent metabolic deterioration and the development of fatty liver diseases.
Recently, administration of nicotinamide riboside, a precursor of NAD+ biosynthesis, has been shown to protect against hepatic fat accumulation in mice fed a high-fat and high-sucrose diet by sirtuin 1 and sirtuin 3-mediated UPRmt, triggering a mitohormetic pathway that enhanced hepatic β-oxidation and mitochondrial complex activity [657]. In addition, a recent study revealed that activating transcription factor 5 is required to maintain mammalian UPRmt during mitochondrial stress, which is essential for organelle recovery [58]. Therefore, further studies to discover the upstream regulators of UPRmt and mitohormesis will provide clues for developing a new class of treatments for fatty liver diseases in humans.
CONCLUSIONS
This review has highlighted the UPRmt and two kinds of mitokines in mammalian systems, which play a protective role in fatty liver and fibrotic liver diseases. However, some important questions remain regarding the mammalian UPRmt and mitohormesis in the development and progression of NAFLD/NASH, alcoholic liver disease, and fibrosis. It would be interesting to determine how UPRmt and mitokines affect systemic energy metabolism, leading to the attenuation of fatty liver and metabolic liver disease. Additionally, we should identify the upstream regulators of UPRmt and mitohormesis in the liver, which might lead to the discovery of drugs for metabolic liver diseases including NAFLD/NASH. Furthermore, several publications have suggested that receptors for mitokines such as GDF15 may be present in peripheral metabolic organs [74750]. Thus, further studies regarding mitokine receptors could provide clues about inter-organ communication by mitohormesis in metabolic liver diseases.
ACKNOWLEDGMENTS
This work was supported by the Korean Endocrine Society of New Faculty Research Award 2018 and Chungnam National University Hospital Research Fund, 2018. Hyon-Seung Yi was also supported by the Basic Science Research Program, National Research Foundation, Ministry of Science and ICT, Future Planning, Korea (NRF-2018R1C1B6004439).
Notes
CONFLICTS OF INTEREST: No potential conflict of interest relevant to this article was reported.