Medical Treatment with Somatostatin Analogues in Acromegaly: Position Statement
Article information
 , Chong Hwa Kim19
, Chong Hwa Kim19
Abstract
The Korean Endocrine Society (KES) published clinical practice guidelines for the treatment of acromegaly in 2011. Since then, the number of acromegaly cases, publications on studies addressing medical treatment of acromegaly, and demands for improvements in insurance coverage have been dramatically increasing. In 2017, the KES Committee of Health Insurance decided to publish a position statement regarding the use of somatostatin analogues in acromegaly. Accordingly, consensus opinions for the position statement were collected after intensive review of the relevant literature and discussions among experts affiliated with the KES, and the Korean Neuroendocrine Study Group. This position statement includes the characteristics, indications, dose, interval (including extended dose interval in case of lanreotide autogel), switching and preoperative use of somatostatin analogues in medical treatment of acromegaly. The recommended approach is based on the expert opinions in case of insufficient clinical evidence, and where discrepancies among the expert opinions were found, the experts voted to determine the recommended approach.
INTRODUCTION
Acromegaly is a chronic disease caused by hypersecretion of growth hormone (GH), and GH-secreting pituitary tumors are found in 95% of acromegaly patients [1]. Acromegaly is a rare disease with an annual incidence of 3.3 cases per million persons, and estimated prevalence of 40 to 130 per million persons [2]. In Korea, the annual incidence was 3.9 cases per million persons in 2007, and the prevalence was 27.0 cases per million persons [3]. The primary treatment for patients with acromegaly caused by pituitary tumors excessively secreting GH is surgery [4] to remove the tumor through a trans-sphenoidal adenomectomy. However, the success rate of surgical treatment varies depending on the size of tumor, extent of tumor invasion and the proficiency of the neurosurgeon performing the surgery [5678]. Approximately 70% of the patients with pituitary microadenomas are able to maintain their serum GH and insulin-like growth factor-1 (IGF-1) concentrations within the normal range after surgery [9]; however, such curative effects can be expected for only 50% of the patients with pituitary macroadenomas [10]. Postoperative medical treatment may be initiated when serum GH concentrations are not reduced to 1 µg/L or lower based on the 75-g oral glucose tolerance test 3 months after surgery, indicating the likely presence of residual tumor [4]. Medical treatment options for these patients include somatostatin analogues (octreotide, lanreotide, and pasireotide), GH receptor antagonists (pegvisomant), or oral dopamine agonists (cabergoline). The treatment goals for acromegaly are to reduce the tumor size and to maintain GH and IGF-1 within normal ranges as well as to improve the clinical symptoms and signs of acromegaly [1]. When surgical treatments are unavailable or are refused by patients, medical treatments may be considered as primary therapy [1].
The Korean Endocrine Society (KES) published clinical practice guidelines for the treatment of acromegaly in 2011. Since then, the number of acromegaly cases, publications on studies addressing medical treatment of acromegaly, and demands for improvements in insurance coverage have been dramatically increasing. In 2017, the KES Committee of Health Insurance decided to publish a position statement regarding the use of somatostatin analogues in acromegaly. Accordingly, consensus opinions for the position statement were collected after intensive review of the relevant literature and discussions among experts affiliated with the KES and the Korean Neuroendocrine Study Group. In this position statement, the recommended approach is based on the expert opinions in case of insufficient clinical evidence, and where discrepancies among the expert opinions were found, the experts voted to determine the recommended approach. The recommendation levels were defined depending upon strength of evidence as shown in Table 1.

Definition of Recommendation Levels
SUMMARY
Indication and treatment goals of using somatostatin analogues in acromegaly
(1) Medical treatment using somatostatin analogues in acromegaly may be initiated when serum GH concentrations are not reduced to 1 µg/L or lower based on the 75-g oral glucose tolerance test 3 months after surgery. Treatment goals are to reduce the tumor size, to maintain GH and IGF-1 within normal ranges and as well as to improve the clinical symptoms and signs (A).
Initial dose and dose adjustment of somatostatin analogues
(1) The initial dose of octreotide long-acting release (LAR) is 20 mg, administered by intramuscular injection at 4-week intervals. If octreotide LAR fails to suppress serum GH concentrations below 2.5 µg/L after the first 3 months of use, the dose may be increased to 30 mg every 4 weeks. The maximum dose is 40 mg every 4 weeks. If serum GH concentrations remained suppressed below 1 µg/L after administering 20 mg octreotide LAR every 4 weeks for 3 months or longer, and the presenting symptoms and signs of disease are alleviated, the dose may be reduced to 10 mg every 4 weeks (B).
(2) Lanreotide autogel (ATG) can be administered at an initial dose of 60, 90, or 120 mg every 4 weeks. If serum GH concentrations are not reduced below 2.5 µg/L after administration of 60 or 90 mg every 4 weeks for 3 months, the dose may be gradually increased to a maximum of 120 mg every 4 weeks (B).
(3) The initial dose of pasireotide LAR is 40 mg every 4 weeks. If serum GH concentrations are not suppressed after the first 3 months of treatment, the dose may be increased to 60 mg every 4 weeks (B).
Switching somatostatin analogues
(1) When one somatostatin analogue is switched to another in order to improve the biochemical profile and reduce adverse effects, the new drug should be administered at the initial dose outlined in Section 2, above (E).
(2) When switching from octreotide LAR to lanreotide ATG, lanreotide ATG can be administered at 60, 90, or 120 mg regardless of the previous dosage of octreotide LAR. When switching from lanreotide ATG to octreotide LAR, the initial dose of octreotide LAR should be 20 mg every 4 weeks (E).
Extended dose interval for lanreotide ATG
(1) For patients whose GH concentrations have been reduced below 2.5 µg/L and clinical symptoms are controlled when administered octreotide LAR or lanreotide ATG for 3 months or longer, the dose interval may be extended to 8 weeks by switching to 120 mg of lanreotide ATG regardless of the dose of somatostatin analogue previously administered (C).
Preoperative use of somatostatin analogues
(1) The routine use of preoperative somatostatin analogues prior to surgical excision of pituitary adenomas is not recommended; however, when surgical treatment is not available or refused, medical treatment may be considered as primary therapy (A).
Special circumstances
(1) Metformin is the preferred treatment for patients who become hyperglycemic during somatostatin analogue therapy, especially with pasireotide LAR. Dipeptidyl-peptidase-4 inhibitors or glucagon-like peptide-1 receptor agonists are also recommended (E).
(2) Use of contraception when somatostatin analogues are administered and discontinuation of somatostatin analogues if pregnancy is considered are recommended (E).
SOMATOSTATIN AND THE SOMATOSTATIN RECEPTOR
The secretion of GH from the anterior pituitary gland is inhibited by many factors including somatostatin from the hypothalamus [11]. Somatostatin is also secreted from the gastrointestinal tract: somatostatin comprised of 14 amino acids is secreted from hypothalamus, and those consisting of 28 amino acids from δ-cells at the duodenum and jejunum [12]. Somatostatin exerts inhibitory effects on all of hormone secretions from the pituitary, pancreas, gut, and gallbladder by binding to somatostatin receptors (SSTR). Five subtypes of the SSTR have been identified [12]: GH-secreting pituitary tumor cells mainly express SSTR type 2 (SSTR2, >95%) and type 5 receptors (SSTR5, >85%) [13], while type 3 (SSTR3) and type 1 receptors (SSTR1) are each expressed on 40% of GH-secreting tumors, and type 4 receptors (SSTR4) are rarely expressed [14]. In addition, SSTR has been confirmed to be expressed within the central nervous system and gastrointestinal tract, and on inflammatory, immune, and endocrine cells [15].
SOMATOSTATIN ANALOGUES
Since the half-life of secreted endogenous somatostatin is only 3 to 5 minutes in vivo, persistent intravenous administration is required in order to maintain therapeutic levels. In addition, the use of endogenous somatostatin may be limited by potential induction of rebound secretion of GH, insulin and glucagon following infusion [16]. As a result, somatostatin analogues with extended half-lives, such as octreotide and lanreotide, have been developed [1718]. Both analogues were developed to lengthen their half-lives while maintain the β-turn structure (tetrapeptide Phe-Trp-Lys-Thr) and the disulfide bridge connecting amino acids (cysteine) at position 3 and 14 of endogenous somatostatin, which plays a critical role in the functions of somatostatin [12].
Octreotide is administered by subcutaneous or intravenous injection. The affinity of octreotide for SSTR2 is 45 times greater than that of endogenous somatostatin (Kd=0.4 pM). The initial dose is 100 to 250 µg with every 8 hours; the dose may be increased to 1.5 mg/day [19]. Its peak plasma concentrations are achieved 40 minutes following administration, and the half-life is approximately 2 hours [1]. While the half-life of octreotide is longer than that of endogenous somatostatin, it must still be administered several times a day, which led to the development of the octreotide LAR formulation, in which octreotide is mixed with a diluent (carboxymethylcellulose sodium, mannitol, water) and microspheres of biodegradable glucose polymer [12]. Octreotide LAR is administered by intramuscular injection, and maximum plasma concentrations are achieved within 1 hour of administration, possibly as a result of drug absorbed to the carrier microspheres [20]. Octreotide LAR is released in a biphasic manner, such that the plasma concentration declines within 12 hours of administration, remains at sub-therapeutic levels for about 7 days, and then increases to reach its peak concentration on day 14 post-injection [21]. Since plasma concentrations remain elevated for about 35 to 60 days and decrease gradually, octreotide LAR is administered at intervals of 4 weeks, and plasma concentrations reach steady state after three injections [20].
Lanreotide with sustained-release (SR) type is another somatostatin analogue formulated as a biodegradable lactide/glycolide copolymer with high affinity for SSTR2 and a half-life of approximately 5 days. Lanreotide SR is administered through intramuscular injection at a dose of 30 or 60 mg every 7 to 14 days [1]. The lanreotide ATG formulation consists of a supersatsupersaturated aqueous solution prepared in a prefilled syringe, which extends the dosing interval to 4 weeks with deep subcutaneous injection [22]. Its half-life is approximately 23 to 29 days with reaching its steady-state after four injections [1].
Based on the previous studies reporting the expression of SSTR5 in tumor cells of GH-producing pituitary adenoma and significant relationships between SSTR5 expression and disease activity in GH-secreting pituitary tumors [2324], the second generation somatostatin analogue with greater affinity for SSTR5 and other SSTR subtypes, named pasireotide, was developed. Pasireotide is a cyclohexapeptide with a molecular weight of 1,047 Da, and the chemical formula of [(2-aminoethyl)aminocarbonyloxy]-L-proline, phenylglycine, and tyrosine (benzyl) [25]. Pasireotide has 40-, 30- and 5-fold greater binding affinity for SSTR5, SSTR1, and SSTR3, respectively, than octreotide or lanreotide, and approximately 2.5 times lower affinity for SSTR2 [26]. Pasireotide LAR with its extended injection interval was developed by using biodegradable polymers similar to those included in the octreotide LAR [12]. Plasma concentrations reach an initial maximum 24 hours after administration, and the concentration declines for the next 7 days, and then increase again to reach a second maximum on day 21 post-injection [27]. Similar to octreotide LAR, plasma pasireotide concentrations reach steady state after three injections when administered at 4-week intervals.
Somatostatin analogues result in inhibition of GH secretion and cell division of somatotropes in the pituitary gland. They also play a role to suppress IGF-1 production in the liver [128]. It has been proposed that somatostatin analogues may reduce tumor size through the apoptosis by induction of ZAC1 protein expression [12], or reduction of the Ki67 index which leads to the decreased cell cycling of GH-secreting pituitary tumor cells [29]. Since the publication of the first case report of normalization of GH and IGF-1 levels and symptoms in acromegaly patients treated with octreotide 30 years ago [30], the reported control rate by using somatostatin analogues vary widely depending on the study conditions. When octreotide LAR was administered at the recommended dose (20 to 30 mg/month), it was reported that IGF-1 and GH were reduced to normal ranges (<2.5 µg/L) among 38% to 85% and 33% to 75% of patients, respectively [31]; similarly, in patients administered lanreotide ATG, GH, and IGF-1 levels were lowered to normal ranges in 39% to 80% and 38% to 80% of patients, respectively [32]. Treatment with octreotide LAR for 48 weeks resulted in significant reductions of tumor size (>20%) in approximately 75% of patients [33], while lanreotide ATG significantly reduced tumor size (>20%) among 54% of patients after 12 weeks [34]. Lanreotide ATG significantly reduced serum GH and IGF-1 concentrations following 6 to 12 months of treatment, according to a retrospective analysis of acromegaly patients in Korea [35]. According to the study with newly diagnosed patients, treatment with pasireotide LAR for 12 months resulted in a greater proportion of patients achieving the treatment goals (GH <2.5 µg/L and IGF-1 within normal range) than treatment with octreotide LAR (31.3% vs. 19.2%) [36]. Subgroup analyses of patients who underwent surgery before medical treatment also produced similar results [36]. A follow-up study found that 48.6% and 45.7% of patients who continued to receive either pasireotide LAR or octreotide LAR, respectively, achieved the treatment goals for up to 12 additional months [37]. Among patients who failed to meet the treatment goal after 12 months and were switched to either pasireotide LAR or octreotide LAR for up to 26 additional months, 17.3% of those switched from octreotide LAR to pasireotide LAR achieved biochemical control, whereas none of the patients switched from pasireotide LAR to octreotide LAR achieved biochemical control [38]. In addition, the pasireotide versus continued treatment with octreotide or lanreotide in patients with inadequately controlled acromegaly (PAOLA) study, which investigated the effects of pasireotide LAR in patients who failed to meet the treatment goals with either octreotide LAR or lanreotide ATG, reported that approximately 20% of patients who were switched to pasireotide LAR 60 mg met the treatment goals [39].
INITIAL DOSE AND DOSE ADJUSTMENT DURING SOMATOSTATIN ANALOGUE TREATMENT
The initial dose of octreotide LAR is 20 mg, which must be administered by intramuscular injection at 4-week intervals. If octreotide LAR fails to reduce serum GH levels below 2.5 µg/L after the first 3 months of treatment, the dose may be increased to 30 mg every 4 weeks. The maximum dose is 40 mg every 4 weeks (Fig. 1). Although it has been recently reported that the mortality rate of patients with acromegaly is similar to that of the general population when GH is reduced below 1.0 µg/L [40], this position statement employs a cut-off GH level of 2.5 µg/L for the changes of drugs and their doses. It is because the accuracy and standardization of the GH assay kits currently available have not yet been established. Moreover, it is recommended that hypersecretion of GH and disease activity of acromegaly not be diagnosed solely based upon serum IGF-1 levels [4142]: IGF-1 secretion can be affected by various factors other than acromegaly, and data on the normal range of IGF-1 concentrations in the Korean population are lacking [41]. Thus, this position statement does not include the IGF-1 level as a criterion to change the drugs and their doses. Once data regarding serum IGF-1 levels in the Korean population become available, IGF-1 will be included in order revisions of this position statement. When the GH level remains suppressed below 1 µg/L after using 20 mg of octreotide LAR every 4 weeks for 3 months or longer and symptoms and signs of excessive GH are alleviated, the dose can be reduced to 10 mg every 4 weeks. The patient's biochemical parameters, clinical signs, and symptoms should be assessed on a regular basis during somatostatin analogue treatment (expert opinion).
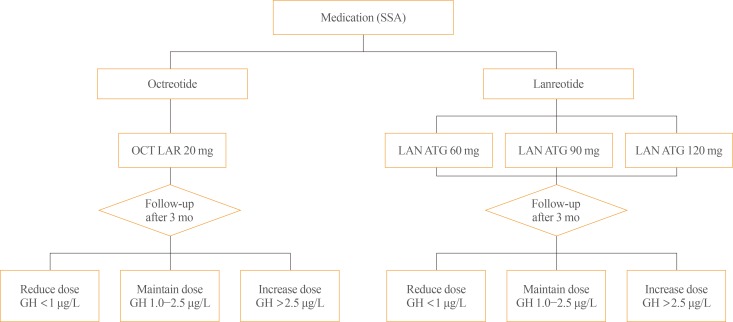
Initial dose and up-/down-titration of octreotide long-acting release (LAR) and lanreotide autogel (ATG) in patients with acromegaly. SSA, somatostatin analogues; OCT, octreotide; LAN, lanreotide; GH, growth hormone.
The initial dose of lanreotide ATG is 60, 90, or 120 mg every 4 weeks. If serum GH levels are not reduced to 2.5 µg/L or less after using 60 or 90 mg every 4 weeks for 3 months, the dose may be gradually increased to a maximum of 120 mg every 4 weeks (Fig. 1). When the GH levels are found to remain suppressed within normal range, the dose may be reduced to 60 mg every 4 weeks, or the 120 mg dose be selected with the dose interval extended to 6 to 8 weeks (refer to the paragraph below for more detailed explanation); in either case, patient symptoms and biochemical parameters should be monitored to verify the continued adequacy of treatment (expert opinion).
The initial dose of pasireotide LAR is 40 mg every 4 weeks. If serum GH levels are not suppressed after the first 3 months, the dose may be increased to 60 mg every 4 weeks.
SWITCHING SOMATOSTATIN ANALOGUES
There is no head-to-head study which has directly compared the therapeutic effects of octreotide LAR and lanreotide ATG during treatment of patients with acromegaly. However, a number of small-scale studies have reported that both therapeutic agents produced similar effects on the improvement of biochemical parameters and reduction of tumor size [43]. Andries et al. [44] reported that both octreotide LAR and lanreotide ATG had almost identical effects on clinical improvement of symptoms and the GH and IGF-1 levels in patients with acromegaly after crossover administration of 10 mg of octreotide LAR and 60 mg of lanreotide ATG, 20 mg of octreotide LAR and 90 mg of lanreotide ATG, and 30 mg of octreotide LAR and 120 mg of lanreotide ATG. In addition, this study found octreotide LAR and lanreotide ATG did not have common adverse effects [44], and it may therefore be beneficial to switch to another somatostatin analogue when one fails to achieve the treatment goals or shows certain adverse effects. In a retrospective study of the effects of those two agents among Korean patients with acromegaly, there was no significant difference with respect to their therapeutic effects [45].
When one somatostatin analogue is switched to another in order to improve biochemical control and reduce adverse effects, the new somatostatin analogue should be administered at the initial doses outlined above (Fig. 2). When switching from octreotide LAR to lanreotide ATG, lanreotide ATG can be administered at either 60, 90, or 120 mg regardless of the previous dose of octreotide LAR; however, when switching from lanreotide ATG to octreotide LAR, the initial dose of octreotide LAR should be 20 mg every 4 weeks. In either situation, serum GH levels should be measured regularly for the first 3 months, and if GH levels are not reduced below 2.5 µg/L, the dose may be increased (expert opinion). When GH secretion is not effectively suppressed after at least 6 months of treatment at the maximum dose of octreotide LAR or lanreotide ATG, the drug may be switched to pasireotide LAR.

Switching between octreotide long-acting release (LAR) and lanreotide autogel (ATG) during the medical treatment of acromegaly.
EXTENDED DOSE INTERVAL FOR LANREOTIDE ATG
The clinical practice guideline for acromegaly published by the Endocrine Society indicates the dose interval may be extended when the treatment goals are achieved with low-dose somatostatin analogues [446]. The Lanreotide Extended ATG Duration (LEAD) study found therapeutic effects were maintained in patients who showed stable disease activity after more than 6 months of treatment with octreotide LAR at 10 or 20 mg every 4 weeks after switching them to lanreotide ATG 120 mg with the dose interval extended to 6 or 8 weeks [47]. Moreover, the extended dose interval (EDI) resulted in the improvement of patients' adherence to treatment by less frequent dosing and reduced drug costs [47]. In a Polish multicenter observational study (the Lanro study), treatment costs were reduced when the dose interval was extended to 8 weeks for patients administered 120 mg of lanreotide ATG [48]. Based on these findings, lanreotide ATG is the only somatostatin analogue of which the extension of the dose interval has been approved for medical treatment of acromegaly [47].
Therefore, for patients whose serum GH concentration is below 2.5 µg/L and clinical symptoms are sufficiently well controlled with octreotide LAR or lanreotide ATG for 3 months or longer, their dose interval may be extended to 8 weeks by switching to lanreotide ATG 120 mg regardless of the dose of somatostatin analogues previously prescribed (expert opinion). The patient's biochemical parameters, clinical signs and symptoms should be assessed on a regular basis during the use of lanreotide ATG with EDI (expert opinion).
PREOPERATIVE USE OF SOMATOSTATIN ANALOGUES
The results of the Preoperative Octreotide Treatment of Acromegaly (POTA) study demonstrated that patients with macroadenomas showed significantly greater reduction of serum IGF-1 concentrations when receiving preoperative octreotide LAR treatment than patients who initially underwent surgical treatments [49]; however, in the follow-up study which investigated these patients for another 5 years, there were no significant differences between the outcomes of the two groups [50]. Thus, the guideline prepared by the Endocrine Society in 2014 does not recommend the routine use of preoperative somatostatin analogues prior to surgical removal of pituitary adenoma for patients with acromegaly [4]. Nonetheless, when surgical treatment is not available or refused, medical treatment may be considered the first-line treatment [1].
SPECIAL CIRCUMSTANCES
It has been reported that the adverse effects of somatostatin analogues may be associated with its inhibition of peristalsis and of pancreatic and gastrointestinal peptide secretion. The adverse effects of somatostatin analogues include nausea, diarrhea, abdominal pain, gastrointestinal paralysis, biliary sludge, and gallstones, but in most cases are mild and can be mitigated by reducing the dose or ceasing administration of the drug [16]. There is also a report of a small number of patients experiencing bradycardia while receiving somatostatin analogs [19]. It has been reported that SSTR2, a major binding receptor of octreotide or lanreotide, is also expressed in pancreatic α-cells [51], and somatostatin analogs do not significantly affect the secretion of insulin in pancreatic β-cells, and rarely results in worsening of glucose control. There are a number of case reports indiindicating glucose control improved as excessive secretion of GH and IGF-1 was resolved [5253]. On the other hand, it was shown that patients administered pasireotide LAR were more likely to experience hyperglycemia or to develop diabetes when compared with those treated with octreotide LAR [36]. Unlike SSTR2 in α-cells, pancreatic β-cells and intestinal L-cells secreting glucagon-like peptide 1 (GLP-1) express SSTR5 to which pasireotide binds with high affinity [1654]. Therefore, insulin secretion was found to be reduced by pasireotide, possibly leading to the deterioration of glucose control. However, insulin sensitivity was not affected [55]. Metformin is the preferred treatment for patients who become hyperglycemic during somatostatin analogue therapy; dipeptidyl-peptidase-4 inhibitors or GLP-1 receptor agonists are also recommended [56].
As for confirmed pregnancy during the use of somatostatin analogues in patients with acromegaly, there have been 50 cases reported with treatment using somatostatin analogues at the time of conception but no malformations have been found in their children [4]. However, it is of note that octreotide can bind to SSTR in the placenta and cross the placenta [57], possibly affecting the fetal development. Reduced blood flow in uterine artery when treated with short-acting octreotide [57] and intrauterine growth retardation after using a lower dose of octreotide LAR [58] have been reported. Due to the limited data available regarding the safety, it is recommended to use contraception when these drugs are administered and to discontinue somatostatin analogues if pregnancy is considered [4]. Considering the prolonged nature of the disease course, interruption of medical therapy using somatostatin analogues for 9 to 12 months in patients with acromegaly should not have adverse effects on the long-term outcome [4].
CONCLUSIONS
Somatostatin analogues may be used when complete recovery cannot be achieved by surgical excision of GH-secreting pituitary adenomas or the patient declines surgery. This position statement is established based on the consensus of opinion among experts and evidence from published data regarding the use of somatostatin analogues in patients with acromegaly. However, this position statement cannot be considered as complete, given the clinical characteristics of acromegaly and the absence of large-scale clinical data in Korea; at this time, the clinical judgement of the physician should take precedence over this statement. This position statement will be revised as needed when additional data for Korean patients become available.
Notes
CONFLICTS OF INTEREST: No potential conflict of interest relevant to this article was reported.
