Lipoprotein Lipase: Is It a Magic Target for the Treatment of Hypertriglyceridemia
Article information

Abstract
High levels of triglycerides (TG) and triglyceride-rich lipoproteins (TGRLs) confer a residual risk of cardiovascular disease after optimal low-density lipoprotein cholesterol (LDL-C)–lowering therapy. Consensus has been made that LDL-C is a non-arguable primary target for lipid lowering treatment, but the optimization of TGRL for reducing the remnant risk of cardiovascular diseases is urged. Omega-3 fatty acids and fibrates are used to reduce TG levels, but many patients still have high TG and TGRL levels combined with low high-density lipoprotein concentration that need to be ideally treated. Lipoprotein lipase (LPL) is a key regulator for TGs that hydrolyzes TGs to glycerol and free fatty acids in lipoprotein particles for lipid storage and consumption in peripheral organs. A deeper understanding of human genetics has enabled the identification of proteins regulating the LPL activity, which include the apolipoproteins and angiopoietin-like families. Novel therapeutic approach such as antisense oligonucleotides and monoclonal antibodies that regulate TGs have been developed in recent decades. In this article, we focus on the biology of LPL and its modulators and review recent clinical application, including genetic studies and clinical trials of novel therapeutics. Optimization of LPL activity to lower TG levels could eventually reduce incident atherosclerotic cardiovascular disease in conjunction with successful LDL-C reduction.
INTRODUCTION
Therapies that lower low-density lipoprotein cholesterol (LDL-C) levels, including statins and proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors, have successfully reduced the risk of atherosclerotic cardiovascular disease (ASCVD) and are currently incorporated in guidelines for the treatment of dyslipidemia [1,2]. Despite this success, recent studies have pointed out that high levels of triglycerides (TGs) and triglyceride-rich lipoproteins (TGRLs) confer substantial residual risk after optimal LDL-C-lowering therapy [3]. Fibrates and omega-3 fatty acids are firstly used or added to treat hypertriglyceridemia in patients whose TG level exceeds 500 mg/dL, which showed high correlation with pancreatitis and lipemia retinalis [4]. Despite the effectiveness of these drugs in lowering TG levels, there have been no other options for patients who do not reach target TG levels than those two, which remains a huge clinical unmet need. Furthermore, inconsistent findings have been reported regarding whether fibrates, omega-3 fatty acids, or TG reduction only can reduce incident ASCVD, with conflicting results from major trials, but still showed benefits in patients with high TG and low high-density lipoprotein cholesterol (HDL-C) [5-8]. However, recent studies, including Asians, have supported the possibility that TG reduction itself may contribute to reducing incident ASCVD [9-11].
In the era of human genomics, it has become possible to discover genetic variants and elucidate their contributions to various phenotypes and diseases. By studying people with hypolipidemia and severe forms of dyslipidemia, such as familial lipid disorders, potential targets for novel lipid lowering therapeutics have been identified, including the LDL receptor, PCSK9, lipoprotein lipase (LPL), the angiopoietin-like (ANGPTL) family, and apolipoproteins (APOs). In fact, evolocumab and alirocumab were developed based on the findings that people with PCSK9 mutations had low LDL-C levels and were protected against ASCVD [12]. With the expansion of new drug development technologies such as small interfering RNA (siRNA), antisense oligonucleotides, and monoclonal antibodies, attempts have been made to test novel therapeutics for TG reduction as well.
In this review, we describe the key modulators and biology underlying the regulation of TGs. LPL is a key regulator of lipolysis that hydrolyzes TGs to glycerol and free fatty acids (FFAs), and ANGPTLs and APOs regulate LPL activity. We describe recent attempts to develop novel therapeutics that alter the function of these TG modulators and clinical trials using those new candidate-drugs.
LIPOPROTEIN METABOLISM AND HYPERTRIGLYCERIDEMIA
Serum TG level is mostly determined by the % of TG core in TGRLs such as chylomicron (CM), very low-density lipoprotein (VLDL) and their remnants lipoprotein particles through lipoprotein catabolism. Thus, the elevation of serum TG in humans is dependent on the abnormalities in production and clearance of TGRLs by primary genetic causes or by secondary diseases such as diabetes, metabolic syndrome, Cushing syndrome, nephrotic syndrome, hypothyroidism, etc. [13,14].
CMs are large sized lipoprotein particles mainly produced from dietary fats which are absorbed through intestinal enterocytes and CM is conjugated with apolipoprotein B48 (APOB48). It can make systemic circulation via lymphatic system and contains large amount of TG core (more than 80% to 90%), which represent high serum TG levels from exogenous pathway. The hydrolysis of CM are mainly dependent on the activity of LPL, which was controlled by apolipoprotein C2/C3 (APOC2/C3), APOA5, and ANGPTLs action [13].
VLDL is a representative lipoprotein which contains large % of TG core, secreted from the liver in the endogenous pathway of lipoproteins metabolism. Sources of TG in VLDL particle come from the FFA spillover from peripheral lipolysis, de novo lipogenesis, CM remnants, etc. [15]. Apolipoprotein B100 (APOB100) is a main APO of VLDL and VLDL remnants. Microsomal TG transfer protein is also important to transfer TG from cytosol to endoplasmic reticulum in hepatocyte or in enterocytes for VLDL or CM assembly with APOB100 or with APOB48 incorporation. Cholesteryl ester TG transfer protein (CETP) is activated when the production of TGRLs is abundant such as in insulin resistance and the enhanced LPL action generates more TG hydrolysis of TGRLs, producing more remnants particles. It is well known that lesser size TGRL particles than CMs can be transported through endothelium and be seated for making atherosclerotic plaque [16]. Thus, the optimizing lowering TGRLs is important treatment not only for the hypertriglyceridemia related complications such as severe pancreatitis and lipemia retinalis but also the progression of ASCVDs [17].
LIPOPROTEIN LIPASE
Lipases are enzymes that catalyze the hydrolysis of TGs. Lipases exist in different contexts; gastric and pancreatic lipases act in an exocrine manner as a component of digestive juices. Hormone-sensitive lipase and adipose TG lipase are located intracellularly and release FFAs and glycerol into the circulation from neutral TGs deposition in adipose tissue. In contrast, LPL and hepatic lipase are located extracellularly on the endothelial cells of blood vessels (Fig. 1).

Overview of the role of lipoprotein lipase in triglyceride metabolism. De novo lipogenesis from the liver results in the secretion of triglycerides in the form of very-low-density lipoprotein cholesterol (VLDL). Dietary fat is transported from intestine to circulation as part of chylomicrons. The triglycerides in chylomicrons and VLDL are hydrolyzed by lipoprotein lipase (LPL). Chylomicron remnants, intermediate-density lipoprotein (IDL) cholesterol, and low-density lipoprotein (LDL) cholesterol are produced as byproducts. LPL is produced from parenchymal cells, including adipose tissue and muscle. LPL is attached to the cell surface via heparan sulfate proteoglycans (HSPGs). Glycosylphosphatidylinositol-anchored high-density lipoprotein binding protein 1 (GPIHBP1) transports LPL from the cell surface to the vascular endothelium. LPL transported to the vascular endothelium hydrolyzes triacylglycerol (TAG) to diacylglycerol (DAG), DAG to monoacylglcerol (MAG), and MAG to glycerol with free fatty acids (FFAs) being released at each step. Parenchymal cells take up FFAs for storage or use FFAs as fuel.
LPL is a key and essential enzyme for the catabolism of TGRLs. LPL is produced by the parenchymal cells of peripheral organs, secreted, and anchored to heparan sulfate proteoglycans on the cell surface. Glycosylphosphatidylinositol-anchored high-density lipoprotein binding protein 1 (GPIHBP1) transports LPL from the cell surface to the capillary endothelium [18]. LPL transported to the vascular endothelium hydrolyzes TGs from CMs and VLDL in the circulation, producing FFAs and glycerol. Insulin induces LPL expression and activity which lead to the uptake of FFAs into peripheral organs for energy storage and consumption. Therefore, LPL is a key regulator of TG levels that is necessary for peripheral organs to store and consume lipids. Familial chylomicronemia syndrome is a rare genetic disorder estimated to affect 1 to 2 individuals per million and characterized by hypertriglyceridemia, which is caused by mutations in LPL or genes that regulate LPL function which include but are not limited to APOC2, APOA5, GPIHBP1, and lipase maturation factor 1 (LMF1) [19,20].
The activity and/or stability of LPL are regulated by various proteins, including ANGPTLs and APOs (Fig. 2). Loss-of-function mutations in APOC2 and APOA5 inhibit LPL, resulting in severe hypertriglyceridemia [21,22]. In contrast, angiopoietin-like protein 3 (ANGPTL3), ANGPTL4, ANGPTL8, and APOC3 inhibit LPL activity, thereby increasing serum TG levels [23]. The role of each protein in the regulation of LPL activity and circulating TG levels is further discussed below.
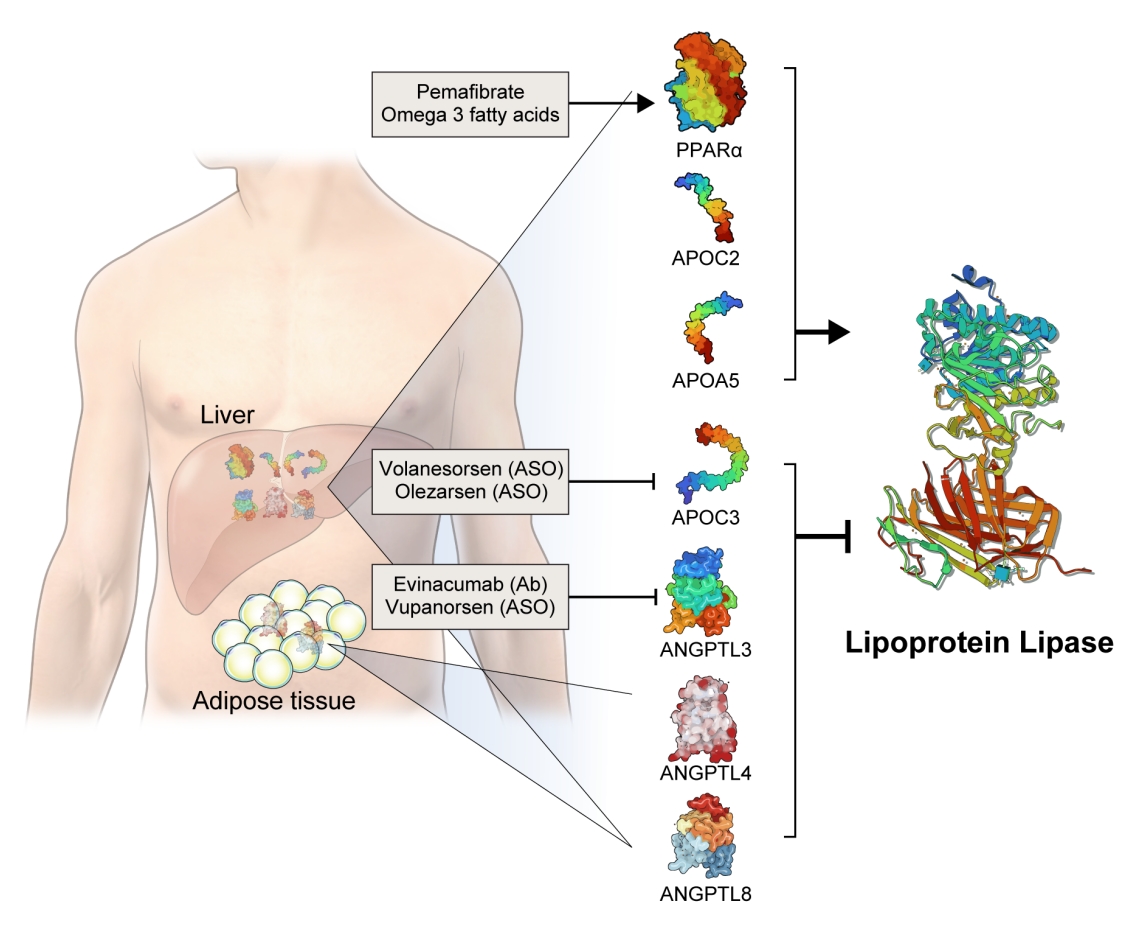
Novel therapeutics that modulate lipoprotein lipase activity. Apolipoproteins (APOs) and angiopoietin-like proteins (ANGPTLs) modulate lipoprotein lipase (LPL) activity: apolipoprotein C2 (APOC2) and APOC5 activate and APOC3, ANGPTL3, ANGPTL4, ANGPTL8 inhibit LPL activity. All of them are produced by the liver, while ANGPTL4 and ANGTPL8 are also produced by adipocytes. Volanesorsen and olezarsen are antisense oligonucleotides (ASOs) targeting APOC3. Evinacumab is a monoclonal antibody (Ab) inhibiting ANGPTL3. Vupanorsen is an ASO targeting ANGPTL3. Peroxisome proliferator-activated receptor alpha (PPARα) activates the transcription of LPL and stimulates LPL activity. Pemafibrate is a PPARα agonist. Omega-3 fatty acids activate both PPARα and LPL.
Based on genetic studies exhibiting altered lipid phenotypes in gain- or loss-of-function mutations in the abovementioned proteins regulating LPL activity, pharmaceutical companies have targeted these proteins to develop novel therapeutics to treat hypertriglyceridemia in patients who do not reach the target goal of TG after using the currently available drugs. The clinical evidence regarding modulation of each protein and the corresponding therapeutic developments are discussed in this review (Table 1).

Clinical Outcomes and Developmental Status of Novel Triglyceride-Lowering Medications Modulating Lipoprotein Lipase
APOLIPOPROTEIN C3
APOC3 is a 79-amino-acid-long small glycosylated protein produced by hepatocytes and enterocytes [24]. APOC3 is a component of TGRLs, and it increases TG concentration by directly inhibiting LPL activity and by preventing the clearance of TGRLs. Genetic studies showed that a heterozygous loss-of-function mutation of APOC3 was associated with a 40% reduced level of plasma TGs and a 40% reduction in incident coronary heart disease [25,26]. APOC3 regulates TG levels via both LPL-independent and LPL-dependent pathways, as APOC3 reduction using the antisense oligonucleotide (ISIS 304801) has shown a TG-lowering effect in people defective for LPL [27]. These genetic studies have established the potential of targeting APOC3 as a therapeutic for lowering TG levels.
Drug development and clinical trials
An antisense oligonucleotide, volanesorsen (previously called ISIS 304801, ISIS-APOCIIIRx, and IONIS-APOCIIIRx) was developed to decrease the level of APOC3 mRNA. Volanesorsen was well tolerated and efficiently reduced APOC3 and TG levels in preclinical and phase 1 studies [28]. In the A Study of Volanesorsen (Formerly IONIS-APOCIIIRx) in Patients With Familial Chylomicronemia Syndrome (APPROACH) trial (NCT02211209), which was a phase 3 trial including 66 patients with familial chylomicronemia syndrome, volanesorsen reduced APOC3 by 84% and TGs by up to 77% at 3 months [29]. However, thrombocytopenia and injection site reactions were common adverse events observed in 45% and 60% of the participants, respectively.
The A Study of Volanesorsen (Formally ISIS-APOCIIIRx) in Patients With Hypertriglyceridemia (COMPASS) trial (NCT02-300233) was another phase 3 trial including 113 patients with fasting TG levels ≥500 mg/dL [30]. At 3 months after treatment, TG levels were reduced by 71%, with an absolute reduction of more than 800 mg/dL. Similar to the APPROACH trial, but to a lesser extent, 24% of patients showed injection site reactions, one patient exhibited thrombocytopenia, and one patient exhibited serum sickness. Based on the positive results from the APPROACH and COMPASS trials, the European Union approved volanesorsen for the treatment of familial chylomicronemia syndrome in May 2019 [31].
To overcome the adverse events observed in volanesorsen trials, an N-acetylgalactosamine-conjugated antisense compound, olezarsen (IONIS-ApoCIII-LRx, AKCEA-APOCIII-LRx or ISIS 678354), was developed to facilitate liver uptake. In a phase 1/2 trial, APOC3 and TG levels decreased by up to 91% and 77%, respectively, at 14 days after a single administration [32]. In a phase 2 trial, TGRLs reduced by 51%, small LDL particles decreased by 39%, and large LDL particles increased by 186% after 6 months of olezarsen treatment suggesting the favorable changes in lipoprotein concentration and particle size remodeling [33]. A phase 3 trial of olezarsen treatment for 53 weeks is underway and is expected to be completed by 2023 (NCT0456-8434).
ANGIOPOIETIN-LIKE PROTEIN 3
Among the eight members of the ANGPTL family, ANGPTL3, ANGPTL4, and ANGPTL8 are responsible for TG regulation by LPL inhibition [34]. The members of the ANGPTL protein family are composed of an N-terminal coiled-coil domain and a fibrinogen-like C-terminal domain, although ANGPTL8 lacks a C-terminal fibrinogen-like domain. The function of ANGPTL3 was first identified in KK/San mice that contained a premature stop codon in Angptl3 and had lower levels of TGs, non-esterified fatty acids, and total cholesterol [35]. Overexpression of Angptl3 or administration of the ANGPTL3 protein restored the features of hypolipidemia.
Human evidence from a genome-wide association study indicated that TG and LDL-C concentrations are affected by genetic variants in ANGPTL3 [23]. Exome sequencing of two patients with familial combined hypolipidemia who did not have an APOB mutation revealed compound heterozygous nonsense mutations of ANGPTL3; these patients exhibited pan-hypolipidemic phenotypes, including low TG and LDL-C levels, with a clear gene-dose association [36].
Drug development and clinical trials
Evinacumab (Evkeeza, Regeneron Pharmaceuticals, Tarrytown, NY, USA) is a human monoclonal antibody inhibiting the action of ANGPTL3 that received approval from the U.S. Food and Drug Administration (FDA) in 2021 as an add-on treatment for patients with homozygous familial hypercholesterolemia who are aged 12 years and older. In a phase 3 trial (the Evinacumab Lipid Studies in Patients with Homozygous Familial Hypercholesterolemia [ELIPSE HoFH]) including 65 patients with homozygous familial hypercholesterolemia whose baseline LDL-C level was 259.5 mg/dL, LDL-C, the primary outcome of this study, was reduced by 47.1% with an absolute change from the baseline of 134.7 mg/dL at week 24 [37]. The mean baseline TG concentration was 97 mg/dL and was reduced by 55.0% at week 24. Adverse events occurred in comparable proportions of patients in the evinacumab and placebo groups (66% and 81%, respectively). An interesting finding was that reductions in LDL-C levels were comparable between those with null-null variants and non-null variants of the LDL receptor. Null-null LDL receptor variants confer higher cardiovascular risk and are less responsive to therapies that depend on LDL receptor activity.
A phase 2 trial that included subjects with or without heterozygous familial hypercholesterolemia who had relatively milder lipid profiles than homozygous patients (baseline LDL-C of 150 mg/dL and TG of 114.5 mg/dL) consistently showed that evinacumab reduced LDL-C by up to 50% and TG by up to 53% [38]. However, the incidence of adverse events and serious adverse events upon evinacumab treatment was up to 80% and 16%, respectively, suggesting that the safety of long-term treatment should be further validated.
A mechanistic study using APOB kinetic analyses showed that evinacumab increased the fractional catabolic rate of APOB for intermediate-density-lipoprotein-cholesterol and LDL-C, suggesting that the LDL-C reduction occurred predominantly by increasing APOB-containing lipoprotein clearance from the circulation [39].
Vupanorsen (AKCEA-ANGPTL3-LRx or IONIS-ANGPTL3-LRx or ISIS 703802) is an N-acetylgalactosamine-conjugated antisense oligonucleotide targeting ANGPTL3. In a preclinical study, vupanorsen reduced levels of hepatic ANGPTL3 and TGs, circulating TG and LDL-C levels, and the progression of atherosclerosis [40]. In a phase 1 study including 44 human participants with TG levels >150 mg/dL, 6-week treatment of vupanorsen reduced TG and LDL up to 63.1% and 32.9%, respectively, without serious adverse events [40]. A phase 2b trial (A Dose-Ranging Study With Vupanorsen [TaRgeting ANGPTL3 with an aNtiSense oLigonucleotide in AdulTs with dyslipidemia, TRANSLATE-TIMI 70]) reached the primary endpoint, achieving a statistically significant reduction in non-HDL-C up to 27.7% at week 24 [41]. Dose-dependent reductions in TG up to 56.8% were observed, while the LDL-C reduction was modest (up to 16.0%). However, adverse events including injection site reactions, elevations in liver enzymes, and a dose-dependent increase in hepatic fat deposition were observed. The magnitude of non-HDL-C and TG reduction was not prominent that the further development of vupanorsen is currently pending. We need to watch for the next movement of this drug.
PEROXISOME PROLIFERATOR-ACTIVATED RECEPTOR ALPHA
Fibrates are agonists of peroxisome proliferator-activated receptor alpha (PPARα) that reduce TG and increase HDL-C levels [42]. The pharmacological actions of fibrates via PPARα agonism activate LPL transcription in the liver and induce fatty acid uptake and β-oxidation [43,44]. PPARα also inhibits APOC3, which may further enhance LPL activity [45]. Transcription of APOA1, APOA2, and APOA5 is induced via PPARα binding [46]. Fibrates further exert anti-inflammatory and anti-atherogenic activity by reducing vascular cell adhesion molecule (VCAM) and monocyte chemoattractant protein-1 (MCP-1) expression via PPRE-dependent and independent manners [47,48].
Previous trials of fibrates showed inconsistent results for preventing ASCVDs [10]. After positive results from the Veterans Affairs HDL-C Intervention trial (VA-HIT) trial, showing a 22% reduction in ASCVD with gemfibrozil (1,200 mg/day) in the 1990s [8], major fibrate trials, including the Lower Extremity Arterial Disease Event Reduction (LEADER) [5], Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) [6], and Action to Control Cardiovascular Risk in Diabetes (ACCORD)-Lipid [7] in the 2000s, failed to demonstrate ASCVD prevention with fibrates. Meta-analyses and Cochrane reviews, in contrast, have suggested that fibrates may contribute to primary and secondary prevention of incident ASCVD [9,49,50]. In particular, patients with high TG and low HDL-C levels benefit most from fibrate treatment [7,51].
Drug development and clinical trials
Pemafibrate is a selective PPARα agonist with improved potency and selectivity compared to fenofibrate. In a phase 3 Japanese trial, the pemafibrate group showed significantly reduced fasting TG levels by 45%, with significant decreases in non-HDL-C and increases in HDL-C levels [52]. In a phase 3 Japanese comparative trial between pemafibrate versus fibrate in patients with baseline TG levels between 300 and 400 mg/dL, the TG-lowering effect of pemafibrate was comparable to that of fenofibrate, with a reduction of up to 50% [53]. Liver and kidney-related adverse events were lower in the pemafibrate group. The Pemafibrate to Reduce Cardiovascular Outcomes by Reducing Triglycerides in Patients with Diabetes (PROMINENT) study was a phase 3 cardiovascular outcome trial including type 2 diabetes patients with TG levels between 200 and 500 mg/dL, but the Kowa Research Institute decided to stop the trial, concluding that the primary endpoint was unlikely to be met without notable safety concerns [54]. Based on their interim analysis and the absence of safety issues, pemafibrate is now considered for other therapeutic applications, including nonalcoholic fatty liver disease. Although pemafibrate failed to prove a preventive effect for ASCVDs, it can be considered for patients with TG levels >500 mg/dL, and it may be safer than fenofibrate.
OMEGA-3 FATTY ACIDS
Omega-3 fatty acids are polyunsaturated fatty acids containing a double bond at the third carbon from the terminal carbon in their chemical structure. Examples of omega-3 fatty acids include α-linolenic acid, which is found in plants, and eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA), which are found in fish. Mammals are unable to synthesize omega-3 fatty acids; therefore, it is essential to consume them through the diet.
It has been suggested that omega-3 fatty acids exert TG-lowering effects via different mechanisms: (1) inducing LPL expression and increasing LPL activity; (2) activating PPARα and δ [55]; (3) suppressing hepatic sterol regulatory element-binding protein 1c (SREBP1c); and (4) increasing the β-oxidation of fatty acids [56]. Six-week supplementation of 6 g of omega-3 fatty acids resulted in a 54% increase in LPL mRNA expression in human adipose tissue and up to a 31% increase in LPL activity [57]. By increasing LPL activity, omega-3 fatty acid supplementation accelerated CM TG clearance [58]. Omega-3 fatty acids were more potent ligands or activators for PPARα than saturated fatty acids [59]. The role of PPARα in LPL and TG reduction is described in the previous section.
Drug development and clinical trials
Omega-3 fatty acids, including EPA and DHA, were shown to reduce TG by 25% to 31% with the administration of 2 to 4 g/day in the EpanoVa fOr Lowering Very high triglyceridEs (EVOLVE) trial [60]. However, cardiovascular outcome trials with different preparations of omega-3 fatty acids have shown inconsistent results. A Study of Cardiovascular Events in Diabetes (ASCEND) and Vitamin D and Omega-3 Trial (VITAL), which administered EPA and DHA in the setting of primary prevention, did not find a significant reduction in ASCVD events [61,62]. The Outcomes Study to Assess Statin Residual Risk With Epanova in High Cardiovascular Risk Patients With Hypertriglyceridemia (STRENGTH) and Omega-3 Fatty acids in Elderly with Myocardial Infarction (OMEMI) trial, which were designed for secondary prevention, did not show a reduction in ASCVD events when a combination of EPA and DHA was administered [63,64]. In contrast, the A Study of AMR101 to Evaluate Its Ability to Reduce Cardiovascular Events in High-Risk Patients With Hypertriglyceridemia and on Statin (REDUCE-IT) trial administered 2 g of a icosapent ethyl, highly purified EPA ethyl ester, and showed a 25% relative risk reduction (17.2% in the icosapent ethyl group vs. 22.0% in the placebo group) in the primary composite endpoint of cardiovascular death, non-fatal myocardial infarction, non-fatal stroke, coronary revascularization, or unstable angina in patients with established ASCVD or with diabetes and other risk factors [65]. This study included subjects who had been receiving statin therapy and achieved LDL-C target levels of LDL-C of 41 to 100 mg/dL. During a median duration of follow-up of 4.9 years, mean fasting TG levels decreased by 21.6% (from 216 to 170 mg/dL), suggesting that high TG levels confer a residual risk of ASCVD after optimal LDL-C lowering therapy, which can be prevented by the administration of EPA.
Although the REDUCE-IT study clearly demonstrated the cardiovascular benefit of EPA, the conflicting results between REDUCE-IT and STRENGTH are in debate based on their study design: active oil (EPA vs. EPA plus DHA); comparator oil (mineral vs. corn); study population (high vs. moderate cardiovascular risk). A post hoc analysis of the Copenhagen General Population Study which mimicked the two trials showed that the contrasting results between the REDUCE-IT and STRENGTH study can partly be explained by a difference in the comparator oil [66]. The mineral oil arm (a comparator oil in the REDUCE-IT study) showed unfavorable changes in LDL-C and C-reactive protein levels resulting in increased ASCVD risk compared to the entire cohort, suggesting that the protective effect of EPA over EPA plus DHA might have been overestimated. This study supports the protective effects against ASCVD in both EPA and EPA plus DHA.
In conclusion, omega-3 fatty acids have shown to reduce TG level and incident ASCVD, but the biological function for different composition of omega-3 fatty acids should be further studied. Adverse events including atrial fibrillation and bleeding should be considered when prescribing omega-3 fatty acids [67].
CONCLUSIONS
In this review, we focused on novel targets for lowering TG levels based on the modulation of LPL activity. Evinacumab, a monoclonal antibody inhibiting ANGPTL3, is approved for use in the United States in patients with homozygous familial hypercholesterolemia. Volanesorsen inhibits APOC3 and was approved in Europe for the treatment of familial chylomicronemia syndrome, but has yet to be approved by the U.S. FDA due to safety issues including thrombocytopenia [68]. Other attempts to enhance LPL activity by inhibiting ANGPTL3/8 complex with monoclonal Ab, showed dose dependent reduction in TG, LDL-C, non-HDL-C in phase 1 trial [69]. We hope more clinical result from on-going studies in near future.
Pemafibrate was found to be effective in lowering TG, with fewer side effects than fenofibrates, but its cardiovascular outcome trial was stopped due to futility issue in ASCVD prevention, but still testing its efficacy in nonalcoholic fatty liver disease. EPA was found to effectively reduce TG levels as well as incident ASCVD, but adverse events, including atrial fibrillation and bleeding, should be kept in mind. Beyond the abovementioned drugs, other therapeutics have been attempted to lower LDL-C with different targets which include inclisiran, bempedoic acid, lomitapide, mipomersen, etc. [70]. But most studies of new lipid lowering treatment were the result on the top of statin therapies and still have very narrow indication for extremely high risk population such as homozygous familial hypercholesterolemia.
For the past 20 years, there have been few convincing novel drugs for dyslipidemia, but many new candidate-drugs have been developed recently. In the field of hypertriglyceridemia treatment, we also had very narrow option in the clinical practice. Understanding the pathophysiological mechanism of TGRLs metabolism and targeting LPL as a key molecule with recent therapeutic approach in the treatment of hypertriglyceridemia can be a very bright signal to optimize patients with intractable TG levels and with high residual risks for ASCVDs. However, it is necessary to understand the mechanisms of action, precise metabolic effects, and possible side effects of these drugs to use them appropriately in clinical practice.
Notes
CONFLICTS OF INTEREST
No potential conflict of interest relevant to this article was reported.
Acknowledgements
This work was supported by the National Research Foundation of Korea funded by the Korea Government (grant number NRF-2018R1A5A2024425 and NRF-2021R1C1C1009875). Special thanks to Dong Su Jang for his effort to illustrate figures.