Updates on Paget’s Disease of Bone
Article information

Abstract
Paget’s disease of the bone is a prevalent bone disease characterized by disorganized bone remodeling; however, it is comparatively uncommon in East Asian countries, including China, Japan, and Korea. The exact cause still remains unknown. In genetically susceptible individuals, environmental triggers such as paramyxoviral infections are likely to cause the disease. Increased osteoclast activity results in increased bone resorption, which attracts osteoblasts and generates new bone matrix. Fast bone resorption and formation lead to the development of disorganized bone tissue. Increasing serum alkaline phosphatase or unique radiographic lesions may serve as the diagnostic indicators. Common symptoms include bone pain, bowing of the long bones, an enlarged skull, and hearing loss. The diagnosis is frequently confirmed by radiographic and nuclear scintigraphy of the bone. Further, bisphosphonates such as zoledronic acid and pamidronate are effective for its treatment. Moreover, biochemical monitoring is superior to the symptoms as a recurrence indicator. This article discusses the updates of Paget’s disease of bone with a clinical case.
INTRODUCTION
Clinical case
A 34-year-old man was referred to the department of endocrinology and metabolism due to elevated blood alkaline phosphatase (ALP) levels (701 IU/L; reference range, 40 to 129). He had fractured his clavicle and finger during childhood. There was no reported family history of bone diseases. He was 167 cm tall and weighed 65 kg, and we could not detect abnormalities during the physical examination. Moreover, he displayed elevated serum osteocalcin (55.0 ng/mL; range, 10.7 to 34.1) and C-telopeptide of type I collagen (CTX; 1.240 ng/mL; range, 0.016 to 0.584) levels. The serum level of calcium, phosphorus, intact parathyroid hormone, and 25-hydroxyvitamin D were normal. We performed clinical evaluation with a plain radiographic skeletal survey and whole-body bone scan in addition to bone densitometry. A plain radiograph of the thoracolumbar spine revealed a mild L3 compression fracture. However, the radiographs of the hand and skull did not reveal any abnormalities. Using a technetium-99m whole-body bone scan, we detected abnormalities in both pelvic bones, cervical vertebrae, T1, T7, T11, L3, and L5/S1 vertebrae, and both the tibiae (Fig. 1A). Bone mineral densitometry coupled with dual-energy X-ray absorptiometry revealed decreased lumbar spine density. How should this patient be managed?
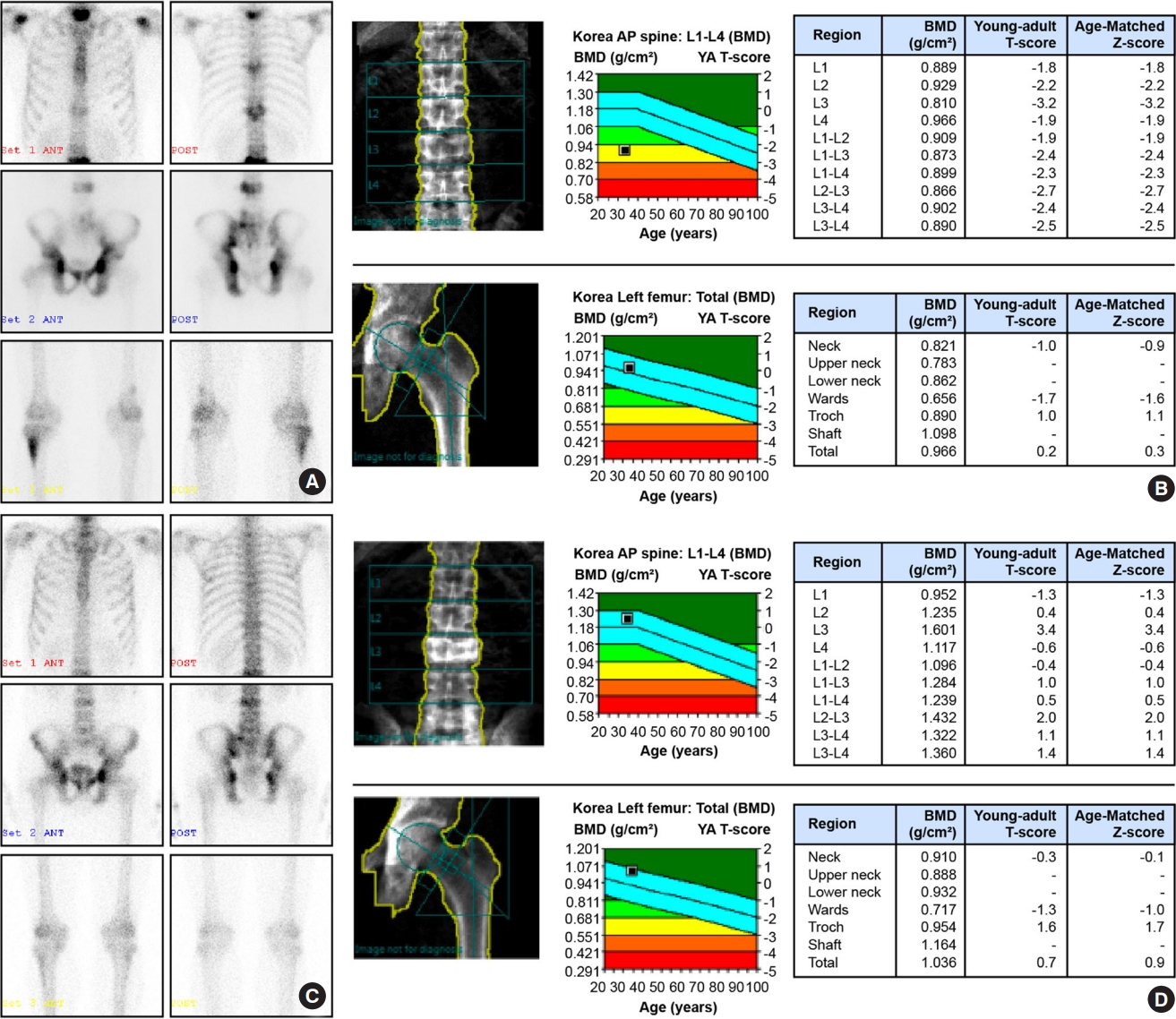
Technetium-99m whole-body bone scans of abnormalities in the pelvic bones, cervical vertebrae, T1, T7, T11, L3, and L5/S1 vertebrae, and both tibial bones. (A) Bone mineral densitometry couple with dual-energy X-ray absorptiometry displaying decreased density of the lumbar spine (B) and improvements in bone abnormalities (C) and density (D) following 10 months of zoledronate administration. AP, anterior-posterior; BMD, bone mineral density; YA, young-adult.
Paget’s disease of the bone (PDB, OMIM 602080) is a chronic and focal bone disorder that typically results in enlargement and deformity in one or more regions of the skeleton [1]. It was first comprehensively described by Sir James Paget, who published his findings in 1877 under the name of “osteitis deformans” [2]. PDB is prevalent in Western countries [3]; however, it is rarely observed in the residents of Asian countries [1]. PDB is associated with defects in bone metabolism that principally include the following clinical manifestations: pain, fractures, and bone tumors owing to overgrowth and deformities in the affected bones [4]. However, there are numerous patients with asymptomatic PDB, and it is predominantly diagnosed incidentally via blood tests that reveal elevated ALP levels or radiological examinations undertaken for other reasons [1]. This review aims to provide updates on the current understanding and the principles of management of PDB.
EPIDEMIOLOGY
In Western countries, PDB is the second most common bone disease, following osteoporosis [5]. It is more prevalent among people with British ancestry. Moreover, it is also prevalent among British immigrants in Australia, New Zealand, North America, and other European nations, including France, Germany, Spain, and Italy [6]. According to archeological analyses of the skeletons, the disease may have originated in England owing to mutations, and subsequently spread throughout Europe and beyond because of the founder effect [7]. PDB is uncommon in African Americans and Asians, particularly those from Korea, China, Japan, and India [8]. There are several single case reports and two case analyses on 11 [9] and eight Korean patients [5] diagnosed with PDB, respectively. A survey of orthopedic surgeons in Japan demonstrated that only 2.8/106 population were affected with PDB [10]. To date, no epidemiological studies have been reported in the Korea [11]. In the United Kingdom (UK), 5.8% of the women and 6.9% of the men aged >85 years are diagnosed with PDB. However, the disease is uncommon in those aged <50 years. It is approximately 1.6 times more prevalent in men than that in women [12] and is prevalent in approximately 2% of the individuals aged ≥55 years in the UK [13]. Moreover, PDB has been less prevalent and severe over the past quarter century in the UK and other nations [14].
CLINICAL MANIFESTATIONS
Bone pain is the most common symptom of those who clinically present PDB. It was reported in 73% of the patients in a recent comprehensive study [15]. Pain mechanisms in PDB are not completely understood. Greater metabolic activity may cause pain in certain people; nonetheless, there is a marginal association between the occurrence of bone pain and metabolic activity in PDB, as measured by the total ALP concentrations. Reid et al. [16] demonstrated that 41.8% of the people with high total ALP did not endure bone pain. In the PRISM (Paget’s disease: randomized trial of intensive versus symptomatic management) research [17], 635 patients exhibited increased ALP at baseline but only 295 (46.4%) patients had bone pain. In addition to discomfort, PDB has other consequences. The majority of clinical characteristics and problems are believed to be caused by disease-specific anomalies in bone remodeling [15]. Hearing loss, basilar invagination of the skull, obstructive hydrocephalus, spinal canal stenosis, and paraplegia may result from the enlarged bones. In a previous systematic review [15], 21.5% of the patients exhibited bone deformity at the initial presentation, followed by deafness (8.9%) and pathological fracture (8.5%). Further, PDB frequently causes osteoarthritis. In 2002, the UK General Practice Research Database demonstrated that patients with PDB were more likely than their age-matched controls to require hip arthroplasty for osteoarthritis (odds ratio, 3.1; 95% confidence interval, 2.4 to 4.1). In cases that require orthopedic surgery, increased vascularity of the bone may result in substantial blood loss. Paraplegia can be seldom caused by vascular “steal” rather than direct spinal cord compression [18]. Increased bone blood flow has been associated with high-output cardiac failure; however, this event is infrequent [19]. The total prevalence of complications in PDB is unclear, since fewer than 10% of the individuals with X-ray evidence for PDB pursue medical help [12]. Osteosarcoma is a rare consequence of PDB and affects approximately 0.3% of the patients [12]. The prognosis is not favorable despite the intensive treatment that has been administered. PDB seldom causes giant cell tumor (GCT). A global literature search identified 117 PDB-related cases of GCT. However, people of Italian ancestry from the Campania region were overrepresented in this series. Notably, several patients with PDB-related GCT in this region have a zinc finger protein 678 (ZNF678) missense mutation [20].
PDB can significantly increase the bone turnover. Both ALP, a diagnostic marker for osteoblastic activity and N-telopeptide of type I collagen (NTX), CTX, markers of osteoclastic activity, are elevated in PDB during both bone formation and resorption [21]. Clinicians can use total serum ALP to measure PDB activity and antiresorptive drug effects. Bone-specific ALP, a procollagen type I N-terminal propeptide (P1NP), is correlated with the disease activity but does not always offer clinical advantage over total ALP in routine practice. However, P1NP levels are a better indication of the disease activity in people with liver disease [22].
PDB radiographs resemble the histological stages. During the osteoclastic phase, which is the earliest stage of the disease, radiographs indicate V-shaped lesions or “blade of grass” appearances in the long bones [23]. In the skull, osteolysis appears as a lucent zone, which is termed as osteoporosis circumscripta. In mixed stages of the disease, this feature may radiologically appear as lysis and sclerosis. The bones may demonstrate osteolysis, coarsening and thickening of the trabeculae, and thickening and broadening of the cortical tissue. PDB can thicken the iliopectineal and ischiopubic lines in the pelvis. Moreover, acetabular protrusion is a common phenomenon. The vertebral bodies may be enlarged throughout the spine, frequently including the posterior components [24]. There may be a “cotton wool appearance” in the skull [25,26]. Sclerosis typically predominates in the terminal stages of an illness. Moreover, a coarsened trabecula pattern persist, as does the bony enlargement. The enlargement of the diploic space in the skull is referred to as a “Tam o’ Shanter” cap, named after the Scottish hat; while in the spine, it is referred to as a “picture frame vertebra.” However, these three histological processes can coexist in one bone. Moreover, radiographs may identify long bone bowing deformities, such as coxa vara of the femoral neck. Stress fractures appear as incomplete fissure fractures on the tension side of the bone along with deformation.
Nuclear bone scans are sensitive to pagetic lesions. Radioisotope bone scans can detect polyostotic involvement. In the early osteolytic stages of PDB, decreased metabolic activity reduces the radioisotope uptake [23].
PATHOLOGY
PDB is characterized by anomalies in all phases of bone remodeling. The fundamental problem comprises the control of osteoclasts, which are enlarged in number and size, and are hypernucleated (up to 50 nuclei per cell). In response to rapid bone resorption, bone production accelerates by 6- to 7-fold, according to tetracycline-based histomorphometric estimations. The new bone formation is disorganized and lacks the normal lamellar pattern; thus, the affected bones are larger and more sclerotic than the normal bones. However, they are of poor quality, which leads to deformity and fracture [27].
In vitro studies of bone marrow cells from the affected bones have demonstrated that pagetic osteoclast precursors have an abnormal “phenotype,” in that they are hypersensitive to osteoclastogenic factors, including receptor activator of nuclear factor kappa Β ligand (RANKL), tumor necrosis factor (TNF), and 1-alpha, 25-dihydroxyvitamin D3—they will form multicellular osteoclasts at concentrations 10- to 100-fold lower than that required for normal osteoclast formation [28-30]. Pagetic osteoclast precursors have higher levels of transcription initiation factor—transcription factor II D (TFIID) subunit 2 (TAFII)-17, which is a component of the TAFIID transcription complex that binds the vitamin D receptor [31]. Other abnormalities in the serum and peripheral blood cells, including elevated interleukin-6 and interferon levels, may be attributed to rapid bone turnover [32].
THE PATHOPHYSIOLOGY OF PDB
Genetics
Genetic factors play a significant role in PDB etiology. Approximately 5%–40% of the patients have a positive family history [29,33-35], and PDB is 7 to 10 times more frequent in the first-degree relatives of the affected patients as compared to controls [35-37]. The disease is inherited as an autosomal dominant trait in several families [38,39]. PDB is caused by a combination of rare, high-penetrance variants in genes, such as sequestosome 1 (SQSTM1), which causes autosomal dominant inheritance, along with common variants in genes such as colony-stimulating factor 1 (CSF1), TNF receptor superfamily, member 11a, and transmembrane 7 superfamily member 4 (TM7SF4), which disrupt the critical proteins that regulate osteoclast differentiation or function, thus causing PDB and related disorders [40,41].
Linkage analysis
Linkage studies in dominantly inherited PDB families identified three susceptibility loci. One locus on 5q35 was separately identified in the French-Canadian and UK populations, while another location on 5q31 was discovered in French-Canadians [39], but it has not been reproduced in other groups. Two additional loci, 2q36 and 10p13, revealed only a suggestive connection with PDB [38]. However, another analysis confirmed a strong linkage with 10p13 and disproved the association with 2q36 [42].
According to cloning studies, mutations in the SQSTM1 gene cause 5q35-linked PDB. A recurrent mutation that changes proline to leucine at codon 392 (P392L) was originally reported in the French-Canadian population [43]. Consequently, further variants in SQSTM1 were identified in the UK population [44] and other groups [34].
SQSTM1
Thirty percent of familial PDB is related to dominantly inherited SQSTM1 mutations [43,44]. SQSTM1 mutation carriers tend to develop the disease at an earlier age and have more extensive illness [45]. Families lacking SQSTM1 mutations exhibit milder familial PDB phenotypes [46]. Patients with truncated SQSTM1 mutations have a more severe phenotype [45]. Approximately 30 SQSTM1 mutations have been associated with PDB [47], despite p.P392L being the most frequent mutation worldwide. The limited number of cases with bi-allelic mutations do not appear to have a more severe phenotype than those with a single mutation. Moreover, 5% of the patients with sporadic PDB have SQSTM1 mutations.
According to a previous report, 80% of the family members inheriting a mutation acquire the illness by 70 years of age [34]. SQSTM1 was identified by positional cloning in families with a high penetrance of the disease; hence, these observations may not be generalizable, and the penetrance may be continuously changing. Some patients may not possess the gene even in families with SQSTM1 mutations, thereby suggesting additional causes [48].
SQSTM1 encodes the signaling-related scaffold protein p62. Despite an unclear mechanism via which p62 alterations cause predisposition to PDB, researchers have principally emphasized on the protein’s effects on osteoclast differentiation, activity, and survival. These effects include nuclear factor kappa B (NF-κB) and oxidative stress-induced Kelch-like ECH-associated protein 1/Nuclear factor erythroid 2–related factor 2 transduction, protein degradation, autophagosome formation, and apoptosis. RANKL-activated osteoclast signaling involves p62. The connection between RANKL and RANK in osteoclasts activates transcription factors, such as NF-κB and nuclear factor-activated T cells c1, which promote and control osteoclast development, activity, and survival [49]. Autophagy removes the damaged proteins and organelles, and p62 facilitates ubiquitin-mediated autophagy (Fig. 2) [50]. The majority of PDB mutations occur at the C-terminal ubiquitin-associated domains [47].
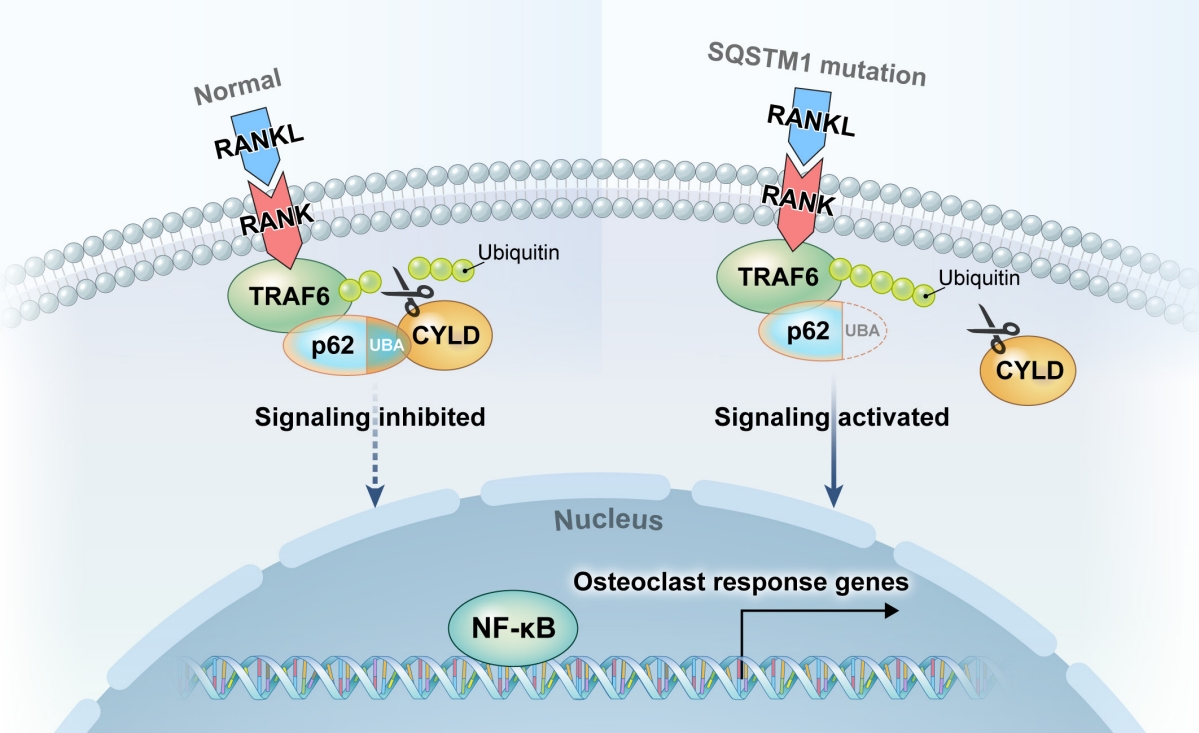
The hypothesized mechanism of osteoclast activation in sequestosome 1 gene (SQSTM1) mutation. Under normal conditions, the ubiquitin-associated (UBA) domain of p62 (light blue) recruits the deubiquitinating enzyme cylindromatosis (CYLD) to the intracellular domain of the receptor activator of nuclear factor kappa B (RANK) receptor, where it deubiquitinates the tumor necrosis factor (TNF) receptor-associated factor 6 (TRAF6) (scissors), thereby inhibiting RANK signaling. Certain mutations of the p62 UBA domain (disrupted lines) inhibit the recruitment of CYLD (yellow), thus leading to increased ubiquitination of TRAF6 (light green) and the activation of RANK signaling. RANKL, receptor activator of nuclear factor kappa Β ligand; NF-κB, nuclear factor kappa B.
Genome-wide association analysis
Genome-wide association studies (GWAS) offer significant insights on the genetic background of PDB. Albagha et al. [40,41] used GWAS to identify seven susceptibility loci in patients with PDB who tested negative for SQSTM1. Strong candidate genes for susceptibility within these loci include CSF1 (1p13), which encodes macrophage colony stimulating factor (M-CSF), a cytokine necessary for osteoclast differentiation [51], RIN3 (14q32) which encode a guanine exchange factor called Ras and Rab interactor 3 [52], optineurin (10p13) which is involved in regulating NF-κB signaling and TM7SF4, which encodes dendritic cell-specific transmembrane protein (DC-STAMP), an important protein for osteoclast precursor fusion [53].
Enviromental factors
In addition to the genetic causes of PDB, researchers have hypothesized potential infection with paramyxoviruses as an alternate etiology. This notion originated from the discovery that osteoclasts in the pagetic bone frequently contain nuclear inclusions that resemble paramyxoviruses. Several studies have reported on the identification of viral mRNA or protein in samples taken from PDB patients utilizing biotechnological amplification methods. However, they were unable to find any similar mRNA or protein in the control samples [54,55]. Several investigations, however, have failed to find viral RNA or protein in PDB patients. Furthermore, no live paramyxovirus from diseased tissues has been identified [56,57].
Furthermore, vitamin D deficiency may possibly play a role as an environmental factor in PDB, since the prevalence of PDB is higher in regions of the UK with prevalent vitamin D deficiency [58]. A low calcium diet and vitamin D insufficiency are thought to worsen severity by causing secondary hyperparathyroidism, which is expected to activate osteoclasts and increase bone turnover. As a result, hyperparathyroidism can aggravate the severity and activity of PDB [27].
In addition, biomechanical factors influence PDB susceptibility, which can be supported by the PDB’s preference for the axial skeleton and weight-bearing extremities. In a patient who was significantly ill with polio, there was no pagetic involvement in the paralyzed leg [59]. Moreover, there have been cases of PDB originating from overused bones [60].
THE DIAGNOSIS OF PDB
Clinicians should use plain radiographs to assess the extent of deformity, detect possible fractures and lytic regions, and analyze the neighboring joints in the bones suspected of PDB [61]. Computed tomography scans and magnetic resonance imaging may benefit in the further evaluation of patients with fracture-related clinical findings but negative radiographs, preoperative planning for arthroplasty or corrective osteotomies, and biopsy planning in patients with Paget sarcoma-related clinical findings [62].
A nuclear bone scan is the most sensitive test for detecting pagetic lesions [61,63]. Repeat radiographs and bone scans are unnecessary unless the patient is experiencing novel or worsening symptoms [63]. It is important to determine the presence of a monostotic or polyostotic disease. In conjunction with a bone scan, clinicians can perform a whole-body radiographic examination to identify the activity and location of the disease [64].
Several markers of high bone turnover can be used for diagnosing and monitoring PDB. Serum ALP is the most commonly used biochemical marker. In asymptomatic patients, increased ALP is frequently the only indicator of disease involvement. Patients with monostotic and, in some instances, polyostotic illness, may exhibit normal or modestly increased serum ALP levels. Sometimes evaluating bone-specific ALP and performing liver function tests is necessary to ensure that the source of eleelevated blood ALP is the bone and not the liver [63].
Patients with PDB have higher levels of P1NP, serum CTX, urine NTX, and urinary hydroxyproline [22,65]. P1NP is often helpful since it is elevated in patients with limited disease, increases early following a recurrence, and responds appropriately to bisphosphonate therapy [22]. Although the majority of PDB patients have normal calcium and phosphorus levels, patients with fractures and restricted mobility may develop hypercalcemia due to increased osteoclast activity [22,65].
Extremely active PDB is associated with accelerated bone formation, which may produce hypocalcemia, particularly in the context of bisphosphonate therapy. This in turn inhibits bone resorption but exerts no effect on bone formation, hence increasing the calcium requirements. Hypocalcemia can cause secondary hyperparathyroidism [22,65,66]. PDB typically manifests in relatively healthy older adults with high ALP, normal blood calcium, normal 25-hydroxyvitamin D levels, and no indication of hepatobiliary disease [15,22,65,66].
Genetic testing
Considering the importance of genetic determinants in PDB and the substantial effect size of risk variants, genetic testing may facilitate identifying patients at a risk of developing PDB or complications [67]. This is particularly important in the uncommon syndromic variants of PDB in which the disease is inherited in a Mendelian fashion, and genetic testing is already undertaken on the children of affected persons. The zoledronate in the prevention of Paget’s (ZIPP) study investigated the possible function of genetic testing coupled with therapeutic intervention. In this study, adults with a family history of PDB underwent genetic testing for SQSTM1 mutations, and those who tested positive were invited to participate in the randomized controlled trial of zoledronic acid or placebo to determine the treatment efficacy in preventing the onset of PDB [68]. Interestingly, the majority of individuals with lesions exhibited normal levels of the biochemical indicators of bone remodeling [69], as determined by an analysis of the baseline features. Guay-Belanger et al. [70] combined a genetic screening test for SQSTM1 mutations with a test for biochemical markers associated with PDB as an alternative method. This method identified 93.9% of the patients with PDB as compared to the control group. Similarly, Albagha et al. [67] used GWAS to assess seven SNPs correlated with PDB in patients who tested negative for SQSTM1 mutations to predict its degree and severity in an international cohort of 1,940 patients. The method effectively identified the disease severity, complications, and the number of bisphosphonate courses administered [67]. The studies by Guay-Belanger et al. [70] and Albagha et al. [67] focused on individuals with PDB, in contrast to those at a risk of PDB owing to a positive family history. Further studies are warranted to investigate the danger of genetic and other markers in predicting the likelihood of PDB.
THE MANAGEMENT OF PDB
The majority of patients have no symptoms and do not require treatment. However, most individuals with active PDB at a risk for further skeletal and extraskeletal problems should be treated with bisphosphonate.
PDB is pharmacologically managed by reducing bone resorption by the osteoclasts. The clinical potency of bisphosphonate is determined by its affinity for hydroxyapatite (which determines the skeletal uptake) and farnesyl pyrophosphate (FPP) synthase inhibition. During bone resorption, the osteoclasts absorb bisphosphonates, which inhibit FPP. This is a vital step in the mevalonate pathway that leads to cholesterol synthesis and generation of geranyl-geraniol that is necessary for intracellular protein prenylation. The disruption of this mechanism causes secondary osteoclast apoptosis [71,72]. Patients without contraindications should be administered a single dose of 5 mg intravenous (IV) zoledronate.
Two clinical trials compared 5 mg IV zoledronate to 30 mg/day risedronate for 2 months. This 6-month study demonstrated that 96% of the patients receiving zoledronate exhibited a therapeutic response, as compared to 74% of those receiving risedronate. ALP was normalized in 89% and 58% of the patients receiving zoledronate and risedronate, respectively. Zoledronate had a faster onset and better quality-of-life outcomes, including bone pain alleviation. Patients who responded to the primary study entered a follow-up study to compare the remission duration of both therapies. At 2 years, 98% and 57% of the patients receiving zoledronate and risedronate, respectively, maintained a therapeutic response, which decreased to 87% and 38%, respectively at 5 to 6 years [71,73,74]. The mean P1NP values were normal in the zoledronate group throughout the follow-up but increased gradually in the risedronate group [74].
Overall, zoledronate displays an excellent safety profile. Flu-like symptoms are the most prevalent adverse effects that occur in approximately 2.5% of the patients. Nonsteroidal anti-inflammatory drugs reduce the frequency and severity of these reactions by approximately 50%. Uveitis and other ocular inflammation can also occur as a part of the acute phase reaction, but their occurrence is rare (1%). Zoledronate can be nephrotoxic, and people with a glomerular filtration rate of 35 mL/min should not take it. Lower doses or longer infusion times may be considered for certain patients, but they are not authorized by the regulatory agencies. In patients with severe vitamin D deficiency (25-hydroxyvitamin D <25 nmol/L), bisphosphonates may cause symptomatic hypocalcemia [75].
In open studies and randomized controlled trials, researchers compared risedronate tablets (30 mg/day for 2 to 3 months) with etidronate. Despite a 73% stabilization of the ALP levels and evidence for pagetic pain alleviation with risedronate, relatively larger doses of oral bisphosphonates exerted considerable upper gastrointestinal adverse effects [76-78]. PDB can be effectively controlled transiently with ibandronate; however, this use has not been extensively recommended [22]. Two randomized trials compared alendronate against placebo and etidronate. Following 6 months of 40 mg/day alendronate administration, 60% to 70% of the patients displayed normalized ALP with healed lytic radiological lesions. Biopsies revealed normal lamellar bone histology [79,80].
Pamidronate normalized bone resorption following 1 week of oral dosage. In contrast, the normalization of bone growth lasted only from 3 to 6 months. IV pamidronate was effective in treating PDB with respect to reduction of biochemical markers, although it was less effective than zoledronate [81].
There are limited data for denosumab, which suppresses osteoclast development and activation. The recurrent subcutaneous application of 60 mg denosumab resulted in the normalization of ALP levels following 4 to 8 months, besides improvement in symptomatic and scintigraphy findings. However, its pharmacological duration necessitates multiple injections to maintain normal bone turnover markers and clinical remission [82-84].
Despite bisphosphonates being the primary treatment for PDB, disease-related complications may necessitate surgical procedures such as joint replacement, osteotomy for deformity, or the surgical management of fractures. Similar rationale exists for surgical procedures in individuals with or without PDB. PDB-associated paraplegia appears to be better controlled with bisphosphonates than that with surgery [72,85-87].
Treatment monitoring
Antiresorptive drugs reduce the resorption markers more rapidly than formation markers. For majority of the patients, assessing the total ALP or other baseline disease activity markers (e.g., CTX) at 6 to 12 weeks is acceptable and cost-effective, particularly during significantly decreased bone turnover. The maximum inhibition of excessive bone turnover may require a 6-month assessment [88].
Patients with osteolytic lesions caused by PDB undergo a second X-ray examination approximately 1 year after the initial radiological diagnosis to establish therapeutic improvement or a deterioration in the absence of therapy. In case of persistent elevation of the biochemical markers of bone turnover or bone pain, clinicians are advised to take additional X-rays to establish resolving lesions. To maximize the duration of remission, the bone turnover marker should be decreased below the midpoint of the reference range for the marker selected for monitoring.
The normal value of pre-pagetic bone turnover is unknown for most patients; therefore, the aim is to reduce the turnover values to the lower half of the reference range. Achieving ALP levels in the lower half of the normal range following zoledronate therapy was associated with a 6-year probability of a therapeutic response loss of 10% [65,74].
The prolonged normalization of bone turnover may prevent or minimize long-term complications such as fractures, deformities, and degenerative joint diseases [87,88]. The measurement of P1NP or total and bone-specific ALP and CTX or NTX is indicated for evaluating the activity of untreated monostotic PDB, despite these values being normal during evident disease activity.
Relapse/Retreat
Biochemical follow-up is a more objective indication of relapse than symptoms in patients with an accelerated bone turnover. The applied treatment agent determines the biochemical monitoring frequency. After achieving normal bone turnover, clinicians should evaluate the persistent response to zoledronate medication every 1 to 2 years. Less potent medications should be monitored every 6 to 12 months [74,87]. Rarely does bone pain reoccur without an increase in bone turnover. Because it is possible that it may be caused by degenerative joint disease, it is an insensitive sign of recurrence [88].
CONCLUSIONS
Clinical case
To determine the molecular diagnosis for the underlying metabolic bone disease, we performed targeted exome sequencing for the candidate genes. We identified a heterozygous variant of SQSTM1, which was previously an unreported nonsense variant in exon 8, c.1273G>T (p.Gly425*; reference sequence: NM_ 003900.4) (Fig. 3). The minor allele frequency of the variant was 0.0004% in gnomAD and 0% in the Korean reference genome database. The patient was diagnosed with PDB and was administered a single dose of IV zoledronate (5 mg). We determined the clinical and biochemical remission of PDB and improvement in bone density. Following 4 and 10 months of zoledronate therapy, his ALP, osteocalcin, and CTX levels decreased to 154 IU/L, 31.5 ng/mL, and 0.471 ng/mL and 100 IU/L, 18.8 ng/mL, and 0.267 ng/mL, respectively. Bone scans revealed reduced abnormalities (Fig. 1A, C) and improved bone density (Fig. 1B, D). The patient experienced fever and a hypocalcemia-related tingling sensation following zoledronate administration. He recovered immediately upon supplementation with calcium, vitamin D, and acetaminophen.

Sanger-sequenced chromatograms for sequestosome 1 gene (SQSTM1) displaying the heterozygous pathogenic variant (c.1237G>T [p.Gly425*]).
PDB is a common bone disease characterized by disordered bone remodeling, but is relatively very rare in East Asia, such as China, Japan, and Korea. Its precise cause remains unknown. It is likely caused by environmental triggers such as paramyxoviral infections in genetically vulnerable people. Increased osteoclast activity increases bone resorption, which recruits osteoblasts and produces new bone matrix. Disorganized bone tissues develop from rapid bone resorption and formation. Increased serum ALP or distinctive radiographic lesions could be the diagnostic clues. Bone pain, bowing of the long bones, larger cranium, and hearing loss are some common symptoms. Radiographic and nuclear bone scintigraphy corroborate the diagnosis. Further, bisphosphonates, such as zoledronic acid and pamidronate, are used for the treatment. Finally, biochemical monitoring is a better indicator of relapse as compared to symptoms.
Notes
CONFLICTS OF INTEREST
No potential conflict of interest relevant to this article was reported.
Acknowledgements
The molecular diagnosis of the patient was supported by grants from the National Research Foundation, Korea (NRF-2017R1C1B5016225; NRF-2019R1F1A1063188). Written consent for publication was obtained from the patient.