Immune Checkpoint Inhibitors and Endocrine Disorders: A Position Statement from the Korean Endocrine Society
Article information
 , Eun Roh2,*, Chang Ho Ahn3,*, Hee Kyung Kim4, Cheol Ryong Ku5, Kyong Yeun Jung6, Ju Hee Lee7, Eun Heui Kim8, Sunghwan Suh9, Sangmo Hong10, Jeonghoon Ha11, Jun Sung Moon12, Jin Hwa Kim13, Mi-kyung Kim,14, The Committee of Clinical Practice Guideline of the Korean Endocrine Society
, Eun Roh2,*, Chang Ho Ahn3,*, Hee Kyung Kim4, Cheol Ryong Ku5, Kyong Yeun Jung6, Ju Hee Lee7, Eun Heui Kim8, Sunghwan Suh9, Sangmo Hong10, Jeonghoon Ha11, Jun Sung Moon12, Jin Hwa Kim13, Mi-kyung Kim,14, The Committee of Clinical Practice Guideline of the Korean Endocrine SocietyAbstract
Immune checkpoint inhibitors (ICIs) including an anti-cytotoxic T-lymphocyte-associated antigen 4 inhibitor, anti-programmed cell death protein 1 (PD-1) inhibitors, and anti-PD-ligand 1 inhibitors are representative therapeutics for various malignancies. In oncology, the application of ICIs is currently expanding to a wider range of malignancies due to their remarkable clinical outcomes. ICIs target immune checkpoints which suppress the activity of T-cells that are specific for tumor antigens, thereby allowing tumor cells to escape the immune response. However, immune checkpoints also play a crucial role in preventing autoimmune reactions. Therefore, ICIs targeting immune checkpoints can trigger various immune-related adverse events (irAEs), especially in endocrine organs. Considering the endocrine organs that are frequently involved, irAEs associated endocrinopathies are frequently life-threatening and have unfavorable clinical implications for patients. However, there are very limited data from large clinical trials that would inform the development of clinical guidelines for patients with irAEs associated endocrinopathies. Considering the current clinical situation, in which the scope and scale of the application of ICIs are increasing, position statements from clinical specialists play an essential role in providing the appropriate recommendations based on both medical evidence and clinical experience. As endocrinologists, we would like to present precautions and recommendations for the management of immune-related endocrine disorders, especially those involving the adrenal, thyroid, and pituitary glands caused by ICIs.
INTRODUCTION
Immune checkpoint inhibitors (ICIs) have emerged as an effective anti-cancer treatment modality [1-3]. ICIs are monoclonal antibodies that block specific cell surface molecules (checkpoints) and restore the ability of T-cells to fight cancer cells [1-3]. Currently, three classes of ICIs have been approved; a cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) inhibitor (ipilimumab), programmed cell death protein 1 (PD-1) inhibitors (nivolumab, pembrolizumab, and cemiplimab), and programmed death-ligand 1 (PD-L1) inhibitors (atezolizumab, avelumab, and durvalumab) [1,2].
Immune checkpoints are crucial for the maintenance of immunological self-tolerance and the protection of tissues from damage when the immune system responds to a pathogenic infection [4]. By changing the immune checkpoint function, however, cancer cells suppress the activity of T-cells that are specific for tumor antigens as one of the immune escape mechanisms [4]. CTLA-4, PD-1, and its ligand PD-L1 are the main targeted immune checkpoints [5]. CTLA-4 is expressed on the cell surface of regulatory T-cells and can inhibit immune activation [2]. PD-1 is a cell surface receptor on activated immune cells and binds two ligands, PD-L1 and PD-L2 [2]. PD-L1 is expressed on cell surfaces of various tissues and cell types, including tumor cells [2]. When activated by one of its ligands, PD-1 inhibits kinases that are involved in T-cell activation [4]. The mechanisms of action of ICIs are demonstrated in Fig. 1.
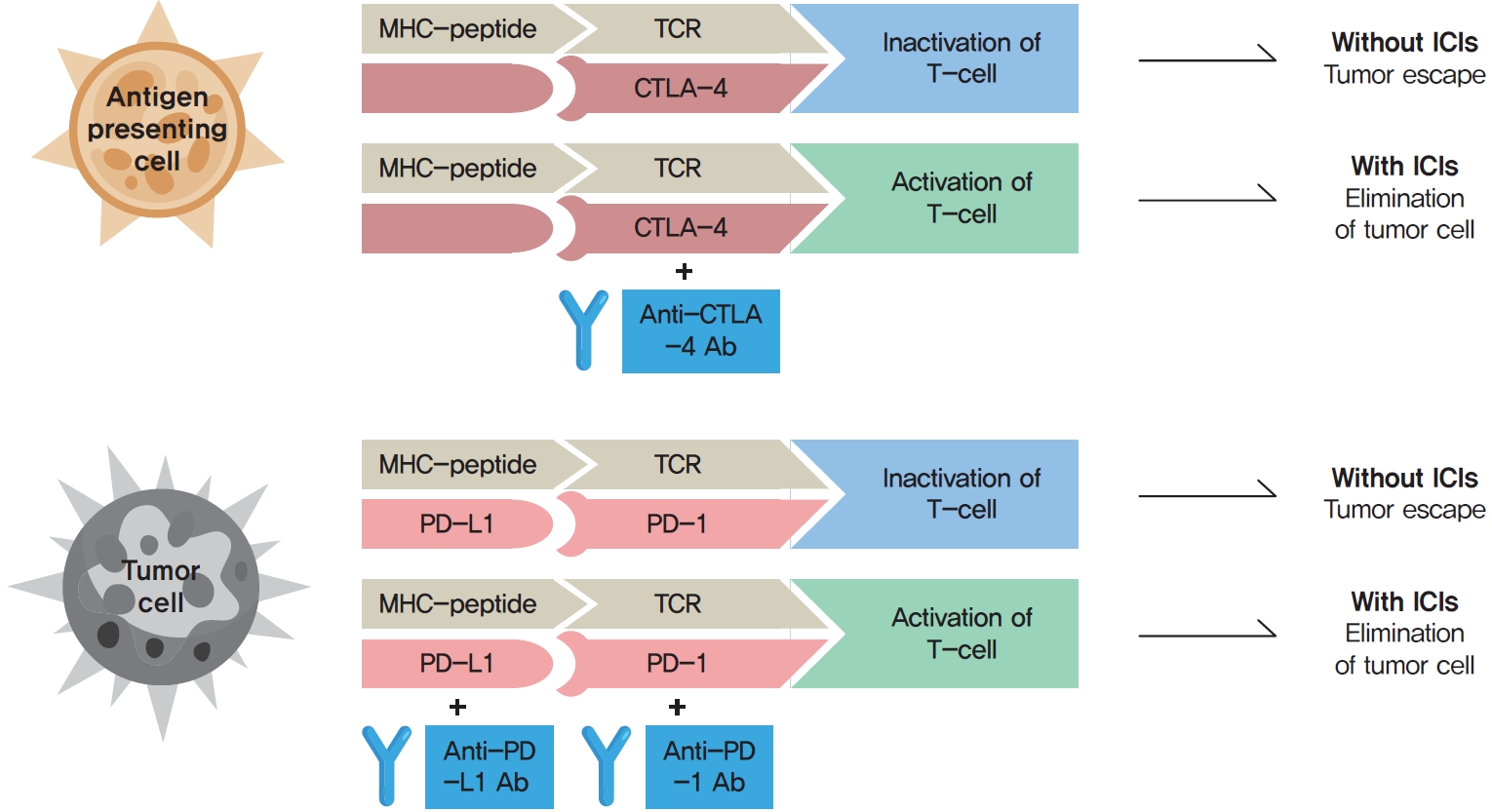
The mechanisms of action of immune checkpoint inhibitors (ICIs). The binding of major histocompatibility complex (MHC)-peptide and T-cell receptor (TCR) activates the anti-cancer pathway of T-cell mediated immune response. Cancer cells suppress the activity of T-cells that are specific for tumor antigens as one of the immune escape mechanisms by changing the immune checkpoint function. The binding of cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) to its ligand and programmed cell death protein 1 (PD-1) to programmed death-ligand 1 (PD-L1) inhibit the anti-cancer activity (tumor escape). The binding of monoclonal antibodies (Abs) to CTLA-4, PD-1, or PD-L1 prevent the inhibition of the anti-cancer pathway, and promote T-cell activation and elimination of tumor cells.
Because immune checkpoints play a crucial role in preventing autoimmune diseases, ICIs can also trigger autoimmune adverse effects, termed immune-related adverse events (irAEs) in the affected organs, including the skin, gastrointestinal tract, liver, lung, and endocrine organs [6]. IrAEs can cause chronic destruction and permanent loss of functions of the affected organ [2,7]. In most cases, irAEs develop in the first few weeks or months of treatment, but they can also develop at any time during ICI therapy, even following cessation of ICIs [2,7]. Endocrine adverse events (AEs) associated with ICIs include hypophysitis, thyroid dysfunction, type 1 diabetes mellitus (T1DM), and adrenal insufficiency [6]. These AEs are a frequent source of acute and persistent morbidity in patients treated with ICIs and can even be life-threatening if not promptly diagnosed and treated [5,7,8].
It has been reported that the incidences of endocrine AEs associated with anti-CTLA-4 and PD-1/PD-L1 inhibitors are different, probably because of their distinct mechanism of action [9]. Anti-CTLA-4 inhibition acts on the triggering stage of the autoimmune process, whereas PD1/PD-L1 inhibition functions in the modulating phase, including peripheral tissues and the neoplastic microenvironment [9]. The high expression of PD-L1/PD-L2 on thyrocytes could be a possible reason [10]. Hypophysitis is commonly related to ipilimumab, one of the anti-CTLA-4 inhibitor, up to 17% of patients [1,9,11-13]. In contrast, hypophysitis associated with anti-PD1/PD-L1 inhibitors is infrequent [1,9,14,15]. Thyroid irAEs, including hypothyroidism and thyrotoxicosis are more common in patients with treated with anti-PD1/PD-L1 inhibitors (0%–40% and 0%–10%, respectively) than in those who receive an anti-CTLA-4 inhibitor (0%–7%), with the highest risk for combined treatment [1,2,9,16]. The overall incidence of ICI-induced T1DM ranges from 0.9% to 2%, and it is mainly associated with anti-PD1 inhibitors [1,2]. Autoimmune adrenalitis is more frequent with the use of PD1/PD-L1 inhibitors than with anti-CTLA-4 inhibitor use, although the incidence rates are low [1,9].
The association between the development of endocrine AEs and oncologic response rates to ICIs remains unclear, and studies are in progress. Recently published studies have demonstrated that ICI-induced endocrine AEs were associated with improved progression-free and overall survival in patients with non-small cell lung cancer, bladder cancer, renal cancer, ovarian cancer, and other types of cancer [17-21].
From a clinical standpoint, the irAEs associated endocrinopathies are important for both clinicians and patients after the initiation of ICIs. In this position statement, we present the classification, screening time points, confirmative tests, and diagnostic and therapeutic approach in patients who develop in irAEs associated with endocrinopathies, with a particular focus on the pituitary, thyroid, and adrenal glands.
PITUITARY TOXICITY
Incidence and etiology
Hypophysitis is one of the most common irAEs, particularly with immune checkpoint blockade involving anti-CTLA-4 inhibition [22]. The incidence of hypophysitis was higher in patients receiving an anti-CTLA-4 inhibitor than in those receiving antiPD-1/anti-PD-L1 antibodies [1,23,24], which may be related to the pituitary expression of CTLA-4 [25]. The incidence of hypophysitis was greatest with combined anti-CTLA-4/anti-PD-1 inhibitor therapy (6.4%) compared with anti-CTLA-4 inhibitor use (3.2%), anti-PD-1 inhibitor use (0.4%), and anti-PD-L1 inhibitor use (<0.1%) [6]. The incidence of hypophysitis induced by anti-CTLA-4 inhibitor use has been found to be related to the drug dose, with more AEs at higher doses [24]. CTLA-4-related hypophysitis typically occurs within several weeks to 3 months after treatment initiation, although it has been reported 19 months after the initiation of treatment [22]. The incidences of pituitary hormone deficiencies of CTLA-4-related hypophysitis has been reported to be 83% for secondary adrenal deficiency, 77% for secondary hypothyroidism, and 53% for hypogonadotrophic hypogonadism [6,26]. Very few cases of posterior pituitary dysfunction have been reported [27,28].
Screening and diagnosis
The symptoms of ICI-related hypophysitis can be nonspecific, including headache and fatigue as the most common presenting symptoms [22,29]. Other symptoms are related to secondary hormone deficiency, such as anorexia, nausea, dizziness, weakness, weight loss, cold intolerance, and decreased libido. Central adrenal insufficiency may be life-threatening if immediate treatment with glucocorticoid is not initiated. Patients with adrenal crisis usually present with an unexplained shock, electrolyte imbalances including hyponatremia, and dehydration. Unlike other forms of autoimmune hypophysitis including lymphocytic hypophysitis, symptoms related to mass effect, such as visual impairment, are rare in ICI-related hypophysitis [27].
If hypophysitis is suspected clinically, a biochemical assessment of the hypothalamic-pituitary axis and imaging of the pituitary gland are recommended. The biochemical assessment of the hypothalamic-pituitary axis includes the adrenal, thyroid, and gonadal axis.
Central adrenal insufficiency is characterized by a low or low-normal cortisol with an inappropriately low or normal adrenocorticotropic hormone (ACTH) level. ACTH and cortisol levels have a diurnal pattern, with early morning levels (e.g., at 8:00 AM) being the most helpful [30]. In normal subjects, baseline levels exceed 5 μg/dL. A cosyntropin (ACTH) stimulation test can help to confirm the diagnosis of adrenal insufficiency. After obtaining a basal cortisol level, 250 μg of intravenous cosyntropin is given and cortisol is tested at 30 and 60 minutes. A peak cortisol level below 18 μg/dL at 30 or 60 minutes after cosyntropin administration is suggestive of adrenal insufficiency [30]. However, a normal cosyntropin stimulation test cannot definitively exclude central adrenal insufficiency of recent-onset, because it takes time for the adrenal glands to atrophy in response to diminished ACTH secretion [31]. The clinical context should be considered to determine whether empirical glucocorticoid replacement is necessary, and close monitoring and repeated testing may be required. A cosyntropin stimulation test also cannot differentiate between hypophysitis and glucocorticoid use, since an exogenous glucocorticoid can suppress corticotropin-releasing hormone (CRH) and ACTH and cause central adrenal insufficiency. These patients should continue glucocorticoid therapy until the glucocorticoid doses can be tapered to a physiological doses. Then, testing for functional recovery of the hypothalamic-pituitary-adrenal axis should be performed before discontinuing the glucocorticoid. In the setting of adrenal insufficiency, increased hypothalamic secretion of CRH stimulates antidiuretic hormone secretion, which results in water retention and hyponatremia [32].
Central hypothyroidism is diagnosed based on a low free thyroxine (fT4) level in the presence of a low to normal thyroidstimulating hormone (TSH) level [30]. A trend of decreasing TSH levels precedes the diagnosis and symptom onset of ICI-associated hypophysitis [33]. It is important to note concomitant nonthyroidal illness syndrome (also called euthyroid sick syndrome) in which similar thyroid function test (TFT) results are present. Nonthyroidal illness syndrome is characterized by a gradual decrease in serum triiodothyronine (T3) levels. A decrease in serum T4 and TSH levels can also be seen if the underlying illness does not improve [34]. The clinical situation should be carefully evaluated, and comparison with baseline TFT results before the initiation of ICI treatment may be helpful.
Hypogonadotropic hypogonadism is characterized by low levels of sex steroids (estradiol in women or testosterone in men) with low or inappropriately normal levels of gonadotropins (luteinizing hormone [LH] and follicle-stimulating hormone [FSH]). Because testosterone levels exhibit a diurnal variation with peak values in the early morning, testosterone concentration should be measured in the morning after an overnight fast [35]. Estradiol levels are low in premenopausal women with hypogonadotropic hypogonadism. Inadequately low FSH and LH levels in postmenopausal women, in whom FSH and LH are typically elevated than in premenopausal women, suggest hypogonadotropic hypogonadism.
The prevalence of growth hormone (GH) deficiency is unclear in ICI-related hypophysitis. In patients with GH deficiency, insulin-like growth factor-1 (IGF-1) levels may be below or within the normal range. However, a normal IGF-I level does not exclude the diagnosis of GH deficiency, making provocative testing mandatory for the diagnosis of GH deficiency [36,37]. However, since GH replacement is contraindicated in patients with active malignancy [36], there is limited utility in testing the GH axis in the setting of ICI-related hypophysitis. Prolactin levels are commonly low in patients with ICI-related hypophysitis [29,33]. Diabetes insipidus were has only been reported in a few cases [27,28].
Pituitary enlargement is a sensitive and specific radiologic feature of ICI-related hypophysitis [33]. Contrast-enhanced magnetic resonance imaging (MRI) scan of the pituitary is the gold standard, and it is pertinent in ruling out metastatic disease. Pituitary enlargement is generally mild to moderate, and pituitary stalk thickening may be seen [33]. Optic chiasm impingement is rare. Radiographic pituitary enlargement may precede biochemical manifestations of hypopituitarism by several weeks [33]. The Society for Endocrinology and Endocrine Emergency guideline recommends an urgent MRI scan of the pituitary gland, particularly if headache, diplopia and cranial nerve palsy are present [38]. In nearly all patients with ICI-related hypophysitis, pituitary enlargement resolves within weeks to months [33,39]. It is possible that radiologic pituitary changes may resolve by the time that biochemical evidence of hypopituitarism is diagnosed. Therefore, a pituitary gland that appears normal on imaging does not exclude hypophysitis, and management should be based on clinical and biochemical evaluation [27].
The package insert for ipilimumab recommends monitoring ACTH and thyroid function prior to ICI initiation and before each dose [40]. ACTH levels within the reference range do not definitively exclude central adrenal insufficiency, especially for patients on ICI therapy (especially ipilimumab-containing regimens) with symptoms or signs of adrenal insufficiency.
Management
Patients with ICI-related hypophysitis may require prompt management, particularly in cases of adrenal insufficiency. High-dose glucocorticoid therapy should be started immediately for patients with adrenal crisis. Intravenous methylprednisolone is only recommended for pressure effects such as optic chiasm impingement, visual field defects, and cranial nerve palsies [38]. Since there is no evidence that high-dose glucocorticoid therapy improves the outcome of ICI-related hypophysitis [39], physiologic doses (5 mg prednisolone once daily or 15 to 20 mg of hydrocortisone in divided doses) are advised. Patients should be counselled with sick day rules and information on stress-dose and emergency glucocorticoid administration [30]. Mineralocorticoids are not required in central adrenal insufficiency, because the renin-angiotensin system is usually preserved [41].
For the treatment of central hypothyroidism, thyroid hormone replacement should be initiated. Importantly, adrenal insufficiency must be excluded before the initiation of thyroid hormone replacement [42,43]. As thyroid hormone replacement can precipitate adrenal crisis in the setting of untreated adrenal insufficiency, glucocorticoid replacement should precede thyroid hormone replacement in cases of concomitant adrenal insufficiency and hypothyroidism. Unlike in primary hypothyroidism, which requires titration of levothyroxine according to TSH, TSH levels are unreliable in central hypothyroidism. Levothyroxine should start at a low dose and be titrated according to fT4 levels to a goal of the mid to upper half of the reference range [34], with TSH monitoring as a signal of recovery of the pituitary thyrotrophs.
Sex hormone replacement may be considered with testosterone for men and estradiol for premenopausal women with ICI-related hypogonadotropic hypogonadism if not contraindicated [30,35]. Testosterone replacement is not recommended in men planning fertility in the near term or in men with breast or prostate cancer, elevated hematocrit, uncontrolled heart failure, recent-onset cardiovascular diseases, or thrombophilia [35]. In men older than 65 years with testosterone deficiency, testosterone therapy may be offered on an individualized basis after a discussion of potential risks and benefits [35]. For women, there are no commonly recognized lists of absolute or relative contraindications to estrogen therapy in professional society guidelines [44]. In general, estrogen therapy should not be used in women with a history of breast cancer or endometrial cancer, a history of venous or arterial thromboembolism including stroke and coronary heart disease, thrombophilia, undiagnosed genital bleeding, liver impairment, or pregnancy [44].
Recovery from ICI-related central adrenal insufficiency appears to be rare. However, recovery from central hypothyroidism and central hypogonadism appears to be fairly common. A previous retrospective cohort study reported that central adrenal insufficiency was persistent at the last follow-up, while the central hypothyroidism resolved in 14 of 22 patients at a median of 10.5 weeks, and hypogonadotropic hypogonadism resolved in seven of 15 patients at a median 15 weeks [39]. Another study reported that adrenal insufficiency remained in 13 of 13 patients, but 11 of 13 recovered from hypothyroidism and 10 of 12 patients recovered from hypogonadotropic hypogonadism at the end of follow-up (median, 33.6 months) [29]. The variations in the reported rates of pituitary hormone recovery may result from differences in the follow-up periods and in strategies for tapering off or testing patients with hormone replacement.
ICI therapy may be resumed in patients with complete or partial resolution after glucocorticoid tapering [40]. The package insert for ipilimumab recommends withholding ipilimumab in the event of symptomatic ICI-related hypophysitis and resuming it in patients with complete or partial resolution of adverse reactions (grade 0 to 1) and who are receiving less than 7.5 mg of prednisone or its equivalent per day. Permanent discontinuation of ipilimumab is recommended in the package insert if there is no complete or partial resolution within 12 weeks of last dose or if it is not possible to reduce prednisone to 10 mg per day (or equivalent) or less within 12 weeks of initiating glucocorticoid therapy. A retrospective cohort study reported that the discontinuation of ipilimumab did not appear to affect the outcome of hypophysitis, and endocrinopathies resolved among a subset of patients who had received prolonged ipilimumab therapy [39].
THYROID TOXICITY
Incidence and etiology
Thyroid dysfunction is the most common endocrine-related irAE-associated with ICIs [6,45]. ICI-induced thyroid AEs include overt or subclinical hypothyroidism and thyrotoxicosis, typically preceding hypothyroidism related to destructive or silent thyroiditis [5,45-47]. Graves’ disease (GD) associated with ICIs has rarely been reported [48,49].
Hypothyroidism is the most frequent endocrine AE with an estimated incidence of 2.5% to 3.8% (for anti-CTLA-4 inhibitor use), 3.9% to 8.5% (for anti-PD1/PDL1 inhibitor use), and 10.2% to 16.4% (for combination treatment), typically occurring at a median time of 8 to 12 weeks after ICI initiation [1,5]. Thyrotoxicosis, typically preceding hypothyroidism, has a lower incidence, which has been estimated to be 0.2% to 5.2% (for anti-CTLA-4 inhibitor use), 0.6% to 3.7% (for anti-PD1/PDL1 inhibitor use), and 8.0% to 11.1% (for combination treatment) [1,5].
Screening and diagnosis
Symptoms and signs suggestive of thyroid irAEs include general weakness, fatigue, palpitation, sweating, anxiety, and weight change. These may appear during the entire course of ICI treatment and even after discontinuation of ICI treatment [2,7]. Because the symptoms and signs associated with thyroid irAEs are nonspecific, it may be difficult to differentiate the symptoms and signs associated with thyroid irAEs from other causes, such as cancer itself or other treatments [50,51]. For this reason, TFTs are essential for an accurate diagnosis, especially in patients with newly developed or progressive symptoms and signs [8].
Baseline TFTs, including TSH, fT4, and total T3, should be performed in all patients prior to the initiation of ICI [2,38,50-52]. It is generally not recommended to screen for thyroid autoantibodies [2,38,50-52]. TFTs should be repeated every 4 to 6 weeks during the first 6 months of ICI treatment, preferably before each treatment cycle starts. The interval can be prolonged to every 2 to 3 months for the next 6 months and every 6 months thereafter.
For patients with hypothyroidism, anti-thyroid peroxidase (TPO) antibody testing is recommended [2,38,50-52]. For the patients with thyrotoxicosis, TSH-receptor antibody testing is recommended to differentiate GD from destructive thyroiditis [2,38,50-52]. Thyroid scintigraphy and ultrasonography are useful for the differential diagnosis in patients with severe or prolonged thyrotoxicosis because some cases of GD have been reported without increased levels of TSH-receptor antibody [53]. Other factors that may affect thyroid function should be considered, such as drugs, nonthyroidal illness, and iodine-based contrast agents [2].
After the cessation of ICIs, regular monitoring of TFTs is needed for at least 2 years [1,2]. Thyroid dysfunction can occur at a later stage, even following ICI cessation and its symptoms are usually nonspecific [1,2,7].
Management
Most of the thyroid AEs are either mild or moderate (grade 1 or 2, according to Common Terminology Criteria for Adverse Events version 5.0) [1,5]. Severe AEs (grade 3 or 4) are rare and found mostly during combination therapy [1,5]. For thyroid irAEs, there is no need to interrupt or stop ICI treatment, except for cases of severe thyroid dysfunction (grade 3 to 4) or severe thyroid eye disease [2,38,50-52].
Patients who have underlying hypothyroidism may need to receive increased doses of levothyroxine after ICI initiation [2]. Hypothyroidism associated with the use of ICIs is often permanent, especially when overt [2]. The goal in the management of subclinical or overt hypothyroidism is to normalize TSH levels by treatment with levothyroxine [2,38,50-52]. Asymptomatic and mild subclinical hypothyroidism (TSH levels of 4.5 to 10 mIU/L) can be monitored without treatment. TFTs should be monitored every 4 to 6 weeks and the levothyroxine treatment should be decided depending on the level and trend of fT4 and symptoms.
Symptomatic, moderate to severe subclinical hypothyroidism (TSH >10 mIU/L), or overt hypothyroidism should be managed with levothyroxine supplementation [2,38,50-52]. A starting dose of 1.6 μg/kg/day in young patients or 25 to 50 μg/day in the elderly or patients with cardiovascular disease is appropriate [2,38,50-52]. Regular monitoring of TFTs is required in order to titrate levothyroxine dosage at intervals of 4 to 6 weeks [2,38,50-52]. Once the TSH is normalized and the levothyroxine dose is adequately titrated, monitoring of TFTs can be extended every 3 to 6 months [2,38,50-52].
In patients with severe symptoms (grade 3 to 4), myxedema, or life-threatening sequale, admission to the hospital and intravenous fluid therapy should be considered [50]. ICIs should be discontinued until symptoms are improved with appropriate levothyroxine replacement [50].
In cases of destructive thyroiditis, an early onset of thyrotoxicosis, which lasts for approximately 6 weeks, is followed by a rapid transition to hypothyroidism requiring long-term levothyroxine replacement [54]. In asymptomatic patients or patients with mild symptoms, TFTs should be monitored every 2 to 3 weeks after diagnosis to observe persistent/progressive thyrotoxicosis or to catch the transition to hypothyroidism [50].
Beta-blockers can be used for the relief of symptoms during the thyrotoxic phase [2,38,50-52]. When thyrotoxicosis persists for more than 6 weeks or GD is clinically suspected, a diagnostic work-up and treatment with anti-thyroid drugs are required [2,38,50-52]. The use of high-dose glucocorticoids for the treatment of thyroiditis is not usually recommended because there is no evidence of its efficacy and there is a risk of secondary adrenal insufficiency (SAI) [55].
In patients with severe symptoms (grade 3 to 4), concern for thyrotoxic storm, or life-threatening sequelae, admission to the hospital and high-dose glucocorticoids therapy should be considered to reduce T4 to T3 conversion [2,50]. ICIs should be discontinued until symptoms are improved [50].
Thyroid eye diseases have been rarely reported and typically developed 2 to 4 months following the initiation of ICI [56]. Patients with severe thyroid eye disease should be referred to an ophthalmologist, and treatment with oral or intravenous high-dose glucocorticoids should be considered [56].
ADRENAL TOXICITY
Incidence and etiology
ICI therapy can induce autoimmune-mediated destruction of the adrenal gland, which leads to primary adrenal insufficiency (PAI). In some cases, imaging evidence of adrenalitis is directly visible on positron emission tomography or computed tomography [6]. In general, ICI-induced complications of the adrenal gland are rarer than those affecting the thyroid or pituitary gland [8]. Due to its rarity, the true incidence of PAI after ICI therapy is not clear. A meta-analysis found that the incidence of PAI was 1.3% to 2.0% for anti-CTLA-4 inhibitor use, 0.8% to 2.0% for anti-PD-1 inhibitor use, and 5.2% to 7.6% for combination treatment [57]. A study analyzing the U.S. Food and Drug Administration Adverse Event Reporting System reported the incidence of PAI after ICI treatment to be 1.03%, which was similar to the results of the previous meta-analysis. However, only 5.5% of all reported PAI cases were classified as confirmed PAI, while the others were considered as suspected PAI [58]. Several clinical trials of ICIs did not report the incidence of PAI and SAI separately. In addition, it is difficult to clearly differentiate PAI from SAI in some cases. If we only consider confirmed PAI, the incidence would be much lower than 1% [6].
Screening and diagnosis
The symptoms and signs suggestive of PAI are similar to those of SAI. Anorexia, fatigue, and general weakness are common symptoms. The characteristic clinical feature of PAI is mineralocorticoid deficiency, which is not seen in SAI. Mineralocorticoid deficiency contributes to more profound hyponatremia, hyperkalemia, metabolic alkalosis, and hypotension [8].
The need for routine screening of PAI in ICI-treated patients is not established. The diagnostic tests for PAI are not different from those of SAI. Measuring morning serum cortisol and ACTH levels can be the first choice of diagnostic test for suspected PAI. An elevated ACTH level with a low morning serum cortisol level (<5 μg/dL) is confirmatory for PAI [59]. However, for those with a morning serum cortisol level over 5 μg/dL, a cosyntropin stimulation test is needed to establish the diagnosis of PAI. Measuring 30 or 60 minutes after the injection of 250 μg of cosyntropin is the standard method, although some centers have adopted a low dose cosyntropin stimulation test using 1 μg of cosyntropin. A peak serum cortisol level below 18 μg/dL is positive for adrenal insufficiency. To differentiate PAI from SAI, an elevated ACTH level is the primary test. In PAI, ACTH is usually elevated to more than twice the upper normal range. However, due to its variability, multiple tests of ACTH may be needed to confirm PAI. Low plasma aldosterone and elevated renin levels or renin activity can be helpful in determining mineralocorticoid deficiency, which is only seen in PAI, not SAI. A priority that should be kept in mind during the diagnostic process of PAI is that empirical glucocorticoid treatment should not be delayed for acutely ill patients with symptoms and signs suggestive of PAI.
Some cases have been found to be positive for autoantibodies to adrenal gland-specific antigens, including 21-hydroxylase [60]. However, the role of adrenal autoantibodies in the pathogenesis, prediction, or prognosis of ICI-induced PAI is not clear [5]. Similarly, adrenalitis is evident on imaging studies in some cases, but it is not universally visible in all cases of PAI. In selected cases, to exclude bilateral adrenal metastasis or other pathology of the adrenal gland, abdominal imaging studies can be performed.
Management
A stress-dose glucocorticoid should be immediately administered to patients with known PAI presenting with an adrenal crisis or severe illness. Hydrocortisone is the preferred choice for a stress-dose glucocorticoid since it has a mineralocorticoid effect. A typical stress-dose of hydrocortisone is an initial 100-mg intravenous infusion, followed by a 50-mg bolus infusion every 6 hours or a 200-mg continuous infusion for 24 hours [5]. Upon patients’ stabilization, the dose of hydrocortisone can gradually be tapered to a basal replacement dose. The regimen of basal glucocorticoid replacement is hydrocortisone (15 to 25 mg per oral in two or three divided doses) [59]. The highest dose should be given in the morning at awakening. The adequacy of glucocorticoid replacement should be determined by a clinical assessment including body weight, postural blood pressure, energy levels, and signs of frank glucocorticoid excess. A mineralocorticoid should also be given to patients with confirmed mineralocorticoid deficiency. Fludrocortisone is given at an initial dose of 50 to 100 μg per day and can be adjusted based on clinical assessments, including blood pressure, serum electrolyte balance, postural hypotension and salt craving [59]. ICI-induced PAI is not usually reversible, necessitating long-term glucocorticoid and mineralocorticoid replacement.
CONCLUSIONS
ICIs targeting immune checkpoints can trigger various irAEs, especially in endocrine organs. IrAE-associated endocrinopathies include hypophysitis, thyroid dysfunction, T1DM, and adrenal insufficiency. Because the symptoms and signs associated with endocrine AEs may be nonspecific but potentially lifethreatening, clinical suspicion and adequate management are important.
CONCISE SUMMARY
Screening
Pituitary
Hypophysitis is the most common pituitary irAE. Patients with symptoms and signs prompting clinical suspicion should be screened for ICI-induced hypophysitis. The symptoms of ICI-related hypophysitis can be nonspecific, including headache and fatigue as the most common presenting symptoms. Symptoms due to mass effect, such as visual impairment, are rare in ICI-related hypophysitis.
Thyroid
Hypothyroidism is the most frequent endocrine AE. Thyrotoxicosis is typically preceding hypothyroidism related to destructive or silent thyroiditis. GD associated with ICIs has rarely been reported. Baseline TFTs should be performed in all patients prior to the initiation of ICI and in patients with newly developed or progressive symptoms and signs suspicious for hyperthyroidism or hypothyroidism.
Adrenal
ICI-induced adrenal toxicity can lead to PAI, which is characterized by deficiency of both glucocorticoid and mineralocorticoid. Patients with symptoms and signs suspicious for PAI should be screened for ICI-induced PAI.
Diagnosis
Pituitary
The biochemical assessment of the hypothalamic-pituitary axis includes the adrenal, thyroid, and gonadal axis. Central adrenal insufficiency is characterized by a low or low-normal cortisol level with an inappropriately low or normal ACTH level. Central hypothyroidism is diagnosed based on a low fT4 level in the presence of a low to normal TSH level. Hypogonadotropic hypogonadism is characterized by low levels of sex steroids (estradiol in women or testosterone in men) with low or inappropriately normal levels of gonadotropins (LH and FSH). Contrast-enhanced MRI scan of the pituitary gland typically shows pituitary enlargement, although a normal scan should not be used to rule out hypophysitis.
Thyroid
TFTs including TSH, fT4, and T3 should be performed. Anti-TPO antibody testing for patients with hypothyroidism and TSH-receptor antibody testing for thyrotoxicosis are recommended.
Adrenal
ICI-induced PAI is diagnosed based on a low morning serum cortisol level or a peak cortisol level <18 μg/dL with elevated ACTH (usually more than a two-fold increase). A low plasma aldosterone concentration with an elevated plasma renin concentration or renin activity supports the diagnosis of PAI.
Management
Pituitary
A high-dose glucocorticoid should be started immediately for patients with adrenal crisis. Physiologic doses of glucocorticoid doses (5 mg of prednisolone once daily or 15 to 20 mg of hydrocortisone in divided doses) are suggested in central adrenal insufficiency, but mineralocorticoids are not required. Thyroid hormone replacement can precipitate adrenal crisis in the setting of untreated adrenal insufficiency; thus, glucocorticoid replacement should precede thyroid hormone replacement. Levothyroxine should start at a low dose and be titrated according to fT4 levels to a goal of the mid- to upper half of the reference range. Sex hormone replacement may be considered, with testosterone for men and estradiol for premenopausal women with ICI-related hypogonadotropic hypogonadism if not contraindicated.
Thyroid
The goal of the management of subclinical or overt hypothyroidism is to normalize TSH levels by treatment with levothyroxine. For thyrotoxicosis, TFTs should be monitored every 2 to 3 weeks after diagnosis in asymptomatic patients or patients with mild symptoms. Beta-blockers can be used for symptom relief during the thyrotoxic phase.
Adrenal
A stress-dose glucocorticoid should be given to patients who are acutely ill and suspected to have PAI. Long-term replacement therapy for PAI includes hydrocortisone daily (15 to 25 mg in two to three divided doses) and fludrocortisone with a starting dose of 50 to 100 μg per day.
Notes
CONFLICTS OF INTEREST
No potential conflict of interest relevant to this article was reported.