Diagnosis for Pheochromocytoma and Paraganglioma: A Joint Position Statement of the Korean Pheochromocytoma and Paraganglioma Task Force
Article information
 , Kyoung Jin Kim2,3,*, Jung Hee Kim4, Mi Kyung Kim5, Chang Ho Ahn6, Kyung Ae Lee7, Seung Hun Lee8, You-Bin Lee9, Kyeong Hye Park10, Yun Mi Choi11, Namki Hong2, A Ram Hong12, Sang-Wook Kang13, Byung Kwan Park14, Moon-Woo Seong15, Myungshin Kim16, Kyeong Cheon Jung17, Chan Kwon Jung18, Young Seok Cho19, Jin Chul Paeng20, Jae Hyeon Kim9, Ohk-Hyun Ryu21, Yumie Rhee2, Chong Hwa Kim22, Eun Jig Lee2
, Kyoung Jin Kim2,3,*, Jung Hee Kim4, Mi Kyung Kim5, Chang Ho Ahn6, Kyung Ae Lee7, Seung Hun Lee8, You-Bin Lee9, Kyeong Hye Park10, Yun Mi Choi11, Namki Hong2, A Ram Hong12, Sang-Wook Kang13, Byung Kwan Park14, Moon-Woo Seong15, Myungshin Kim16, Kyeong Cheon Jung17, Chan Kwon Jung18, Young Seok Cho19, Jin Chul Paeng20, Jae Hyeon Kim9, Ohk-Hyun Ryu21, Yumie Rhee2, Chong Hwa Kim22, Eun Jig Lee2
Abstract
Pheochromocytoma and paraganglioma (PPGLs) are rare catecholamine-secreting neuroendocrine tumors but can be life-threatening. Although most PPGLs are benign, approximately 10% have metastatic potential. Approximately 40% cases are reported as harboring germline mutations. Therefore, timely and accurate diagnosis of PPGLs is crucial. For more than 130 years, clinical, molecular, biochemical, radiological, and pathological investigations have been rapidly advanced in the field of PPGLs. However, performing diagnostic studies to localize lesions and detect metastatic potential can be still challenging and complicated. Furthermore, great progress on genetics has shifted the paradigm of genetic testing of PPGLs. The Korean PPGL task force team consisting of the Korean Endocrine Society, the Korean Surgical Society, the Korean Society of Nuclear Medicine, the Korean Society of Pathologists, and the Korean Society of Laboratory Medicine has developed this position statement focusing on the comprehensive and updated diagnosis for PPGLs.
SUMMARY
1. New classification of pheochromocytoma/paraganglioma
1.1. All pheochromocytoma and paraganglioma (PPGLs) are considered to have metastatic potential. The terms “benign” and “malignant” should not be used to distinguish non-metastatic PPGLs from metastatic PPGLs (A).
1.2. The tumor-node-metastasis (TNM) staging system should be evaluated in diagnosing PPGLs (A).
2. Biochemical tests of pheochromocytoma/paraganglioma
2.1. Initial biochemical tests of PPGL should include measurements of plasma free metanephrines or urinary fractionated metanephrines (A).
2.2. Measurements of urinary dopamine and plasma 3-methoxytyramine are useful for the biochemical diagnosis of PPGLs with predominantly dopamine secretion and/or high risk for metastases (C).
2.3. Chromogranin A can be used as a biomarker for biochemically silent PPGL (PPGL with normal metanephrine, normetanephrine, and 3-methoxytyramine) (C).
3. Imaging of pheochromocytoma/paraganglioma
3.1. Once here is clear biochemical evidence of a PPGL, anatomic imaging by computed tomography (CT) is the first-choice imaging modality to locate PPGLs. Magnetic resonance imaging is the second-choice imaging method when CT findings are inconclusive or when patients are poor candidates to undergo contrast-enhanced CT (A).
3.2. Functional imaging is recommended for evaluating disease characteristics and detecting metastases, particularly in patients with a high-risk for metastases and multifocal diseases (e.g., lager tumor size >5.0 cm, extra-adrenal, bilateral, or hereditary) (A).
3.3. We suggest 123I-metaiodobenzylguanidine (MIBG) scintigraphy/single-photon emission computed tomography (SPECT), gallium 68 (68Ga) 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA)-somatostatin receptor analogs (SSA) positron emission tomography (PET)/CT, or fluorine-18-L-dihydroxyphenylalanine (18F-DOPA) PET/CT as the functional imaging modality according to the genotype, location, availability of radiopharmaceuticals, and clinical situation (B).
3.4. In PPGL patients planning for 131I-MIBG treatment, 123I-MIBG is necessary for treatment decision and response monitoring. 68Ga-DOTA-SSA PET/CT is necessary in patients with metastatic PPGLs when peptide receptor radionuclide therapy (PRRT) is planned (B).
4. Pathological grading system of pheochromocytoma/paraganglioma
4.1. The Pheochromocytoma of the Adrenal Gland Scaled Score (PASS) and the Grading of Adrenal Pheochromocytoma and Paraganglioma (GAPP) cannot be used to confirm the diagnosis of malignancy (B).
4.2. The loss of succinate dehydrogenase B (SDHB) protein by immunohistochemistry staining in tumor cells is suggested to detect the presence of germline mutations in one of the SDHx genes. PPGLs associated with SDHB mutation have a high risk of metastases (B).
5. Genetic testing for pheochromocytoma/paraganglioma
5.1. Genetic testing is recommended in all patients diagnosed with PPGLs (A).
5.2. Genetic testing should be also considered for first-degree relatives of patients with hereditary PPGLs (B).
5.3. Validated targeted next-generation sequencing (NGS) is a preferred method for the genetic diagnosis of PPGLs (B).
5.4. We recommend targeted NGS panels of gene sets based on the current level of evidence of their pathogenic driver status: 10 basic panel (fumarate hydratase [FH], myc-associated protein X [MAX], neurofibromatosis 1 [NF1], rearranged during transfection [RET], succinate dehydrogenase A [SDHA], SDHB, SDHC, SDHD, transmembrane protein 127 [TMEM127], von Hippel-Lindau [VHL]) and five extended panel (egl-9 family hypoxia inducible factor 1/prolyl hydroxylase domain 2 [EGLN1/PHD2], endothelial PAS domain-containing protein 1 [EPAS1], kinesin family member 1B [KIF1B], receptor tyrosine kinase [MET], succinate dehydrogenase complex assembly factor 2 [SDHAF2]) (C).
INTRODUCTION
Pheochromocytomas and paragangliomas (PPGLs) are rare chromaffin cells-derived tumors that originated from the adrenal medulla and the extra-adrenal sympathetic or parasympathetic paraganglia, respectively. The overall annual incidence of PPGLs is estimated to be two to eight cases per million, and the recent report in Korea showed the annual incidence of 1.8 cases per million [1–4].
PPGLs release catecholamines, mainly norepinephrine and epinephrine, causing hypertension, headache, sweating, and palpitations. If not recognized, PPGLs can severely affect the cardiovascular, gastrointestinal, and other systems, and can threaten patients by causing such as fatal arrhythmia, myocardial infarction, cerebrovascular events, and sudden death [5–7]. PPGLs have the potential for metastases, in the case of metastatic PPGLs, found in the presence of tumors derived from chromaffin cells in non-chromaffin organs at the time of diagnosis or during the follow-up period [8]. According to a recently published study, metastases were already detected in 9.0% of patients with PPGLs at the initial diagnosis, and 9.5% of metastases occurred during the follow-up period [1]. Excess of catecholamine, as well as metastases involve in increased morbidity and mortality in PPGLs patients [9–11]. Therefore, timely and accurate diagnosis and treatment of PPGLs is essential.
In recent years, with the advancement of next-generation sequencing (NGS) technology, the genetic discoveries of PPGLs have increased significantly, and at least 30% of these tumors have been identified as hereditary [12]. Over the past decade, our knowledge in the field of PPGLs has been rapidly expanded and changed by many discoveries in genetics, biochemical, imaging diagnosis, and treatment of these tumors. Therefore, the modifications in the fourth edition of the World Health Organization (WHO) classification in 2017 is primarily based on the novel findings on the clinical behaviors and genetics of adrenal tumors [13]. There have been exponential advances in the genetics of these tumors according to the progress in NGS technology, and in recent years, new susceptible genes with germline and somatic mutations have been discovered [12,14,15]. In addition to catecholamines, various products secreted by PPGLs, such as chromogranin A, have begun to be applied as useful biochemical diagnostic markers [16]. Beyond the traditional functional imaging study with 123I-metaiodobenzylguanidine (MIBG) scintigraphy, the emerged molecular imaging techniques, such as 11C-hydroxyephedrine positron emission tomography/computed tomography (PET/CT) and gallium 68 (68Ga) 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA)-somatostatin receptor analogs (SSA) PET/CT, have been introduced for the precise diagnosis for localization and staging [17,18].
In accordance with the revised international recommendations and advanced diagnostic techniques, the PPGL task force team has developed the guideline for the diagnosis of PPGLs, regarding controversial issues in South Korea. The following will discuss the changes noted in the new WHO classification and provide updates on genetic, biochemical, and imaging diagnostic approaches of the PPGLs.
METHODS
Development of evidence-based recommendations
This guideline was developed by the multidisciplinary committee, which comprises the Korean Endocrine Society, the Korean Surgical Society, the Korean Society of Nuclear Medicine, the Korean Society of Pathologists, the Korean Society of Radiology, and the Korean Society of Laboratory Medicine. The guideline included the most current evidence-based recommendations for diagnosis of PPGLs. The grading system included the following considerations: numbers of well-designed randomized controlled trials, meta-analysis results, cohort studies, patient–control studies, or expert opinion on clinical experiences. The guideline committee’s grading system uses A, B, C, or E to present the evidence level supporting each recommendation, as defined in the previous study (Table 1) [19].

Summary of Strength of Evidence for Recommendations
DISCUSSION OF THE RECOMMENDATIONS
1. New classification of pheochromocytoma/paraganglioma
Substantial changes have been included regarding the topics of adrenal tumors in the fourth edition of the WHO classification of endocrine tumors published in 2017 compared to the third edition of 2004 [20,21]. In the 2017 version of the WHO classification, information on ‘tumors of the adrenal medulla and extra-adrenal paraganglia’ has been described in an independent chapter and has been separated from the chapter discussing the ‘tumors of the adrenal cortex’ [20]. Table 2 summarizes the WHO classification of tumors of the adrenal medulla and extra-adrenal paraganglia [20].

Updated Version of 2017 World Health Organization Classification of Tumors of the Adrenal Medulla and Extra-Adrenal Paraganglia
1.1. All PPGLs are considered to have metastatic potential. The terms “benign” and “malignant” should not be used to distinguish non-metastatic PPGLs from metastatic PPGLs (A)
Prior to the update on adrenal tumors in 2017 WHO of endocrine tumors, PPGLs have traditionally been classified as “malignant” and “benign” based on the presence of distant metastases. Metastases occur in approximately 5% to 10% of pheochromocytomas [8]. Several relevant indicators based on histopathology, immunohistochemistry (IHC), genetic mutations and molecular biological characteristics such as the Pheochromocytoma of the Adrenal Gland Scaled Score (PASS) grading system, the Grading of Adrenal Pheochromocytoma and Paraganglioma (GAPP), and the composite pheochromocytoma/paraganglioma prognostic score (COPPs) scoring system have been proposed to predict metastatic potential [22–26]. However, these scoring systems is not well-validated for the prediction of the metastatic tumors. Due to the lack of the approved histological system on PPGL’s biological aggressiveness, all PPGLs are considered to have metastatic potential. Therefore, the terms “malignant” and “benign” are abandoned and combined into a single section “pheochromocytoma” in the updated version of WHO classification of endocrine tumors [20]. In addition, in the current version of the WHO classification, “malignant” was replaced with the term “metastatic” to clearly distinguish between locally invasive and distant metastatic PPGLs.
1.2. The TNM staging system should be evaluated in diagnosing PPGLs (A)
The first staging system for PPGL was published by the American Joint Committee on Cancer (AJCC) in 2017. The AJCC TNM (T, size and location of primary tumor; N, regional lymph node metastases; and M, presence and location of distant metastases) staging system helps to make treatment and prognostic related decisions and provides standardized communicative description tools for the tumor. The TNM classification system for PPGLs reflects the prognostic factors that predict the metastases and shorter survival, and these predictors include the large size of the primary tumor (≥5.0 cm), extra-adrenal tumor, and the presence of distant metastases (e.g., bone, liver, lungs, and lymph nodes) (Table 3) [27]. While smaller (<5.0 cm) tumors rarely occur distant metastases, patients with larger (≥5.0 cm) tumors have lower overall survival rates due to the increased risk of distant metastases [28,29]. Therefore, pheochromocytomas are divided into T1 (<5.0 cm) and T2 (≥5.0 cm) according to the tumor size. Tumors with the invasion of surrounding tissues such as liver or kidneys are classified as T3 because they need extensive surgery and usually tend to be more aggressive [30]. Also, the location of the primary tumors is an important predictive factor of metastases. Sympathetic paragangliomas (PGLs) are associated with higher metastatic potential and shorter overall survival rates regardless of the primary tumor size, so these are reflected in T staging [29]. Patients with metastatic PPGLs have high morbidity and mortality rates due to the excessive catecholamines, and patients with metastatic PPGLs have a significantly lower 5-year survival rate compared to patients without metastases (90% vs. 60%) [31,32]. Similarly, a recent nationwide population-based epidemiological study in South Korea showed the hazard ratio for mortality of patients with metastatic PPGLs was twice higher compared to patients without metastatic PPGLs [1]. The common metastatic sites of metastatic PPGLs are the regional and distant lymph nodes, bone, liver, and lungs [33,34]. Among common metastatic sites, bone metastases are known be less aggressive with a longer median overall survival (12 years) compared with non-skeletal metastases (5 to 7.5 years) [35]. Given that the median survival rates differ according to the metastatic sites, M staging of AJCC was determined by the location of the distant metastases [32].

The AJCC Staging System of Pheochromocytoma and Paraganglioma
2. Biochemical tests of pheochromocytoma/paraganglioma
2.1. Initial biochemical tests of PPGL should include measurements of plasma free metanephrines or urinary fractionated metanephrines (A)
Evidence of catecholamine excess is an essential prerequisite for the clinical diagnosis of PPGL. Metanephrines are the O-methylated metabolites of epinephrine and norepinephrine. They are produced within the cytoplasm of adrenal chromaffin cells or PPGL tumor cells by catechol-O-methyltransferase. This process occurs continuously and independently from exocytic catecholamine release, which is the theoretical basis of superior sensitivity of measuring metanephrines than catecholamines for the diagnosis of PPGL.
Previous clinical studies consistently reported superior sensitivity and specificity of metanephrines than other biochemical tests for PPGL. One of the early studies, which included 214 pheochromocytoma patients from National Institutes of Health (NIH) and European centers, demonstrated a sensitivity of 99% and specificity of 89% for plasma free metanephrines [36]. The diagnostic performance of plasma free metanephrines was significantly higher than urinary catecholamines or vanillylmandelic acid based on the receiver operating curve analysis. A meta-analysis of the diagnostic accuracy of plasma free metanephrines estimated the pooled sensitivity and specificity as 97% and 94% [37].
Several studies compared the diagnostic accuracy of urinary fractionated metanephrines and plasma free metanephrines. Plasma free metanephrines showed a higher sensitivity and specificity than urinary fractionated metanephrines [36,38–41]. However, the difference was small and not statistically significant. Currently, no priority was recommended between plasma free metanephrines and urinary fractionated metanephrines.
The diagnostic accuracy of metanephrines was also evaluated in Korean PPGL patients. In a single-center study of 28 patients and 156 control subjects, plasma free metanephrines showed sensitivity and specificity of 96% and 76%, respectively. Urinary fractionated metanephrines showed sensitivity and specificity of 96% and 94%, respectively [42]. Another study enrolled patients from two large Korean centers also reported high diagnostic accuracy of urinary fractionated metanephrines [43].
For accurate measurement and interpretation of metanephrines, measurement methods, sampling conditions and cut-offs of the assay should be considered. Liquid chromatography with mass spectrometric or electrochemical detection methods is highly accurate and reproducible with low risk of interference. The position of blood sampling should be considered when interpreting the results of plasma free metanephrines. Previous studies used blood sampling in the supine position. Upright positioning stimulates norepinephrine release and subsequently increases plasma normetanephrine. Seated sampling generally reported higher cut-off for diagnosing PPGL and lower specificity than supine sampling [44]. Thus, supine position is recommended for blood sampling, especially when retesting after equivocal elevation of metanephrines.
2.2. Measurements of urinary dopamine and plasma 3-methoxytyramine are useful for the biochemical diagnosis of PPGLs with predominantly dopamine secretion and/or high risk for metastases (C)
As metanephrines are metabolites of epinephrine and norepinephrine, 3-methoxytyramine is O-methylated metabolite of dopamine produced by catechol-O-methyltransferase. Plasma 3-methoxytyramine level can be elevated in PPGL patients due to the excess production of dopamine. The diagnostic accuracy of plasma 3-methoxytyramine for the diagnosis of overall PPGL is not superior to metanephrines. The addition of plasma 3-methoxytyramine to plasma free metanephrines did not significantly increase the diagnostic accuracy for PPGL [45]. However, for some PPGLs that produce dopamine predominantly, plasma 3-methoxytyramine can provide superior diagnostic utility than metanephrines. Plasma 3-methoxytyramine was elevated in dopamine-producing PGLs by nearly 100-folds, suggesting high discriminative power of plasma 3-methoxytyramine for dopamine-producing PPGLs [46].
Elevated plasma 3-methoxytyramine is associated with a high risk of metastatic PPGL. In a large European cohort of 365 PPGL patients, 105 patients with metastatic PPGL showed significantly higher plasma 3-methoxytyramine and urinary dopamine [47]. The mean plasma 3-methoxytyramine level was higher by nearly 4-folds in metastatic PPGL than non-metastatic PPGL. In addition, succinate dehydrogenase B (SDHB) mutant PPGL showed increased production of dopamine in the tumor metabolomics and had high plasma and urinary dopamine and plasma 3-methoxytyramine levels [48]. SDHB mutation confers a high risk of metastases, which, at least partly, explains the high risk of metastases in PPGL with elevated dopamine and 3-methoxytyramine.
2.3. Chromogranin A can be used as a biomarker for bio-chemically silent PPGL (PPGL with normal metanep-hrine, normetanephrine, and 3-methoxytyramine) (C)
Chromogranin A serves an important role in the formation of secretory granules of neuroendocrine cells. Plasma or serum chromogranin A level is elevated in patients with neuroendocrine tumors. Chromogranin A is highly sensitive and specific for PPGLs when differentiating PPGL patients from normal individuals [49,50]. Chromogranin A was still elevated in biochemically silent PPGLs with normal metanephrine, normetanephrine and methoxytyramine [51]. The histologic evaluation in silent PPGLs showed preserved secretory granule without the expression of tyrosine hydroxylase, which explains the molecular mechanism of elevated chromogranin A in biochemically silent PPGLs.
3. Imaging of pheochromocytoma/paraganglioma
3.1. Once there is clear biochemical evidence of a PPGL, anatomic imaging by CT is the first-choice imaging modality to locate PPGLs. MRI is the second-choice imaging method when CT findings are inconclusive or when patients are poor candidate to undergo contrast-enhanced CT (A)
For the localization of PPGLs, both CT and magnetic resonance imaging (MRI) are highly sensitive. CT with contrast provides an excellent detection rate between 88% and 100% [52–54]. Considering that most PPGLs are located in the abdomen, a CT scan of the abdomen and pelvis should be the first anatomical imaging modality. CT is preferred to MRI for the detection of lung metastases, while MRI shows better sensitivity between 90% and 95% for detecting skull base and neck PGLs than CT [55,56]. MRI is also preferred in patients with an allergy to CT contrast agent, and in patients in whom radiation exposure should be limited (i.e., children, pregnant women, patients with known germline mutations, and those with recent excessive radiation exposure) [57].
3.2. Functional imaging is recommended for evaluating disease characteristics and detecting metastases, particularly in patients with a high-risk for metastases and multifocal diseases (e.g., lager tumor size >5.0 cm, extra-adrenal, bilateral, or hereditary) (A)
3.3. We suggest 123I-MIBG scintigraphy/scintigraphy/ single-photon emission computed tomography (SPECT), 68Ga-DOTA-SSA PET/CT, or Fluorine-18-L-Dihydroxyphenylalanine (18F-DOPA) PET/CT as the functional imaging modality according to the genotype, location, availability of radiopharmaceuticals, and clinical situation (B)
In patients with a high-risk for metastatic disease, such as large size of the primary tumor, extra-adrenal, multifocal (except skull base and neck PPGLs), and recurrent disease, the use of functional imaging modalities are suggested for detecting metastases. 18F-fluorodeoxyglucose (FDG) PET has been used for this purpose; however, other functional imaging is currently recommended as the first choice of image because of its higher sensitivity and specificity [58]. The reported diagnostic performance of each functional modality is different according to disease location, disease extent, and genotype (Table 4).

Diagnostic Performance of Functional Imaging Modalities for Pheochromocytoma and Paraganglioma
123I/131I-MIBG scintigraphy has a comparable sensitivity for pheochromocytoma to fluorine-18-L-dihydroxyphenylalanine (18F-DOPA) PET/CT and 68Ga-DOTA-SSA PET/CT [59–62]. The diagnostic performance is enhanced with use of 123I-MIBG and single-photon emission computed tomography (SPECT) [62,63]. However, the sensitivity of 123I-MIBG scintigraphy in PGLs and metastatic PPGLs is relatively suboptimal, particularly for SDHx-related PPGLs [64–67].
68Ga-DOTA-SSA PET/CT is the most sensitive tool for detecting PPGLs in patients with unknown genetic status based on a recent systemic review and meta-analysis [68]. The elevated clinical value of 68Ga-DOTA-SSA PET/CT is also observed in extra-adrenal PGLs and head and neck PGLs [69–71]. Furthermore, 68Ga-DOTA-SSA PET/CT is preferred in the metastatic PPGLs [72]. In PPGLs with underlying SDHx mutations, 68Ga-DOTA-SSA PET/CT can be recommended [73]. In contrast to SDHx-related PPGLs, 18F-DOPA PET/CT showed high tumor uptake in von Hippel-Lindau (VHL), rearranged during transfection (RET), neurofibromatosis 1 (NF1), myc-associated protein X (MAX), hypoxia-inducible factor (HIF) 2A, prolyl hydroxylase (PHD)1/2, and fumarate hydratase (FH) mutated PPGLs [74,75]. The exact mechanism for this phenotype is currently largely unrevealed, and many of these reports are based on extremely rare cohorts.
Despite the clinical relevance of each functional imaging modality according to disease characteristics and genotypes, the practical availability of radiopharmaceuticals and clinical situation should be considered. 68Ga is produced by a generator system that is affordable only in large centers, whereas 123I/131I-MIBG, 18F-FDG, and 18F-DOPA are commercially available in many countries including Korea. Additionally, all of the functional imaging modalities are effective despite relative inferiority to another in a specific situation. Therefore, it is recommended to appropriately utilize the recommended first choice or alternative functional imaging modalities that are available for each institution (Fig. 1).

Proposed clinical algorithm for nuclear (molecular) imaging studies for pheochromocytoma/paraganglioma. PPGL, pheochromocytoma/paraganglioma; PHEO, pheochromocytoma; HNPGL, head and neck paraganglioma; SDH, succinate dehydrogenase; VHL, von Hippel-Lindau; RET, rearranged during transfection; NF1, neurofibromatosis 1; MAX, myc-associated protein X; HIF2A, hypoxia-inducible factor 2A; PHD1/2, prolyl hydroxylase domain 1/2; FH, fumarate hydratase; 123I-MIBG, 123I-metaiodobenzylguanidine; 68Ga-DOTA-SSA, gallium 68 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid-somatostatin receptor analogs; 18F-DOPA, fluorine-18-L-fluorodihydroxyphenylalanine; 18F-FDG, 18F-fluorodeoxyglucose; PRRT, peptide receptor radiotherapy.
3.4. In PPGL patients planning for 131I-MIBG treatment, 123I-MIBG imaging is necessary for treatment decision and response monitoring. 68Ga-DOTA-SSA PET/CT is necessary in patients with metastatic PPGLs when PRRT is planned (B)
In patients with metastatic PPGLs, targeted radiopharmaceuticals treatment may be considered. In 123I-MIBG scintigraphy-positive metastatic PPGLs, high dose 131I-MIBG therapy can be beneficial [76]. In particular, high-specific-activity 131I-MIBG (Azedra, Progenics Pharms Inc., New York, NY, USA) has received U.S. Food and Drug Administration approval for metastatic or locally aggressive PPGLs, which showed radiographic response rate of 92% and biochemical response of 68% in the phase-II study [77]. Pre- and post-treatment 123I/131I-MIBG is essential to select appropriate candidate and response monitoring. In patients who are planning peptide receptor radionuclide therapy (PRRT), 68Ga-DOTA-SSA PET/CT should be performed. 177Lu, 90Y, 225Ac-labeled DOTA-D-Phe1-Tyr3-octreotide (TOC) and DOTA-0-Tyr3-octreotate (TATE) is the currently available PRRT as compassionate or off-label use [78,79]. Currently, 177Lu-DOTA-TATE is commercially available in many countries including Korea, although it has not been officially approved for PPGL in most countries due to lack of clinical trials. The overall disease control rate of PRRT is 84%, and the biochemical response rate is between 61% and 64% in a meta-analysis [79]. Thus, it is recommended to consider PRRT in patients with metastatic PPGL who are not eligible for conventional chemotherapy and 131I-MIBG, when effective uptake is observed on 68Ga-DOTA-SSA PET/CT.
4. Pathological grading system of pheochromocytoma/paraganglioma
4.1. The PASS and the GAPP cannot be used to confirm the diagnosis of malignancy (B)
To predict risks for PPGL metastases, two separate histological prediction systems have been established as followed; the PASS grading system and the GAPP (Table 5) [25,26]. First, the earliest grading system for detecting the potential of metastatic behavior of pheochromocytoma was the PASS grading system created by Thompson [26] in 2002. PASS originally designed for pheochromocytomas only incorporates 12 individually histologic features endowed with different points (one or two points), which were based on the occurrence of parameters in a pre-defined metastatic cohort. With a total score of 20 of PASS, a pheochromocytoma is considered as biologically aggressive when applying a cut-off score of ≥4 whereas those with a score <4 are considered not to have metastatic potential [26,80]. The PASS grading system has been validated in previous studies, of which the results were inconsistent [81–83]. These studies generally indicate that the PASS system is of high sensitivity (100%) and low specificity with the potential to correctly “rule in” metastatic pheochromocytomas [58,84]. However, this system also carries its shortcomings. First, it has inter- and intra-observer variations [33]. Second, other important factors such as gene mutation, tumor characteristics, and clinical characteristics of patients were ignored, whereas only histological characteristics were included [58,84]. These advantages have been debated in terms of the clinical value of the PASS grading system, which limited application to all PPGL cases for clinical prognostication [80]. Then, the GAPP was developed by Kimura et al. [25] for both PPGLs, which included some histological features from the PASS and added immunohistochemical (Ki-67 index) and biochemical (catecholamine type) data. The GAPP model stratifies the PPGLs into three classes: well-differentiated (0–2 points), moderately differentiated (3–6 points), and poorly differentiated (7–10 points) [25]. Like the PASS system, the GAPP model also has a high sensitivity (90%) and low specificity [58]. Wachtel et al. [85] recently published a retrospective cohort study, including 143 subjects with PPGLs about the comparison PASS and GAPP scoring system. They found that a higher GAPP score was associated with metastatic PPGLs, whereas the PASS score was not. According to their study, PASS had low-moderate reliability between observers, while the GAPP score had significantly less inter-observer variability than PASS. For these reasons, they recommended that a shift from PASS to GAPP scores to predict more accurate prognostication for PPGL patients, but both of them were still insufficient because neither of them was associated with mortality from PPGL. None of them is 100% predictive for risk stratification, but both of them display excellent sensitivity [84].
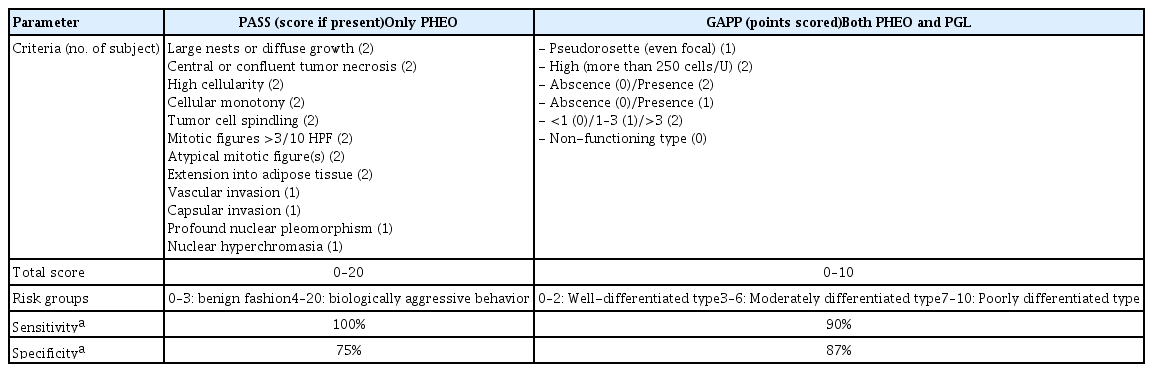
Comparison of Two Systems Predicting Pheochromocytoma and Paraganglioma Metastatic Potential
4.2. The loss of SDHB protein by IHC staining in tumor cells is suggested to detect the presence of germline mutations in one of the SDHx genes. PPGLs associated with SDHB mutation have a high risk of metastases (B)
Pathogenic germline variants in any of the SDHx subunit genes (SDHA/B/C/D) have been implicated in hereditary tumorigenesis of PPGLs [86]. The majority of patients with hereditary PPGL syndromes (30% to 40%) have germline mutations in these SDHx subunit genes [86]. Germline mutations of SDHB have been widely accepted as a high-risk factor for metastases, leading to metastatic PPGLs in 40% or more of affected patients [57,87,88]. Furthermore, SDHB germline mutation is a poor prognostic factor in patients with metastatic PPGLs [11]. In terms of the 5-year probability of survival after the diagnosis of the first metastases were 0.36 (0.15 to 0.57) in SDHB mutation carriers and 0.67 (0.47 to 0.81) in the absence of SDHB mutation (relative risk, 2.6; P=0.019) [24]. SDHB mutation carriers also have a shorter median survival of 42 months, compared to 244 months for SDHB mutation non-carriers. This shorter median survival was significantly associated with the presence of SDHB mutations independent of age at diagnosis.
Germline mutations in any of these SDHx genes lead to destabilization of the SDH protein complex and loss of SDHB expression at IHC. Therefore, loss of SDHB IHC staining in tumoral tissue suggests the presence of germline mutations in one of the SDHx genes with 100% sensitivity and 84% specificity [80,89,90]. Several studies have reported that a combination of biochemical, histological, and SDHB IHC might be useful for predicting metastatic PPGLs [24,25]. One of them was the modified GAPP (M-GAPP) proposed by Koh et al. [24] in 2017. Furthermore, SDHB IHC test is technically easy and cost-effective because of its simplicity of the standard procedure and data interpretation. Hence, it can be routinely performed in all PPGLs, even in the absence of familial or clinical indications for a specific form of inherited PPGL. Therefore, it is necessary to identify SDHx (SDHB, SDHC, and SDHD) germline mutation by the loss of SDHB IHC staining in all patients with PPGLs [91].
5. Genetic testing for pheochromocytoma/paraganglioma
5.1. Genetic testing is recommended in all patients diagnosed with PPGLs (A)
PPGLs have striking characteristics regarding the importance of genetic testing, which are their high heritability and genetic heterogeneity [92]. PPGLs are the most frequently heritable tumors in humans, with at least 30% to 40% of inherited forms of the disease [93]. For this reason, it is recommended that all patients are ‘engaged in shared decision making’ and ‘considered’ for genetic testing, respectively [57,94]. There are several reasons genetic testing should be considered in all patients with PPGLs. First of all, more than one third of all patients with PPGLs have a disease-causing germline mutation in PPGL susceptibility genes because of their strong genetic determinism [95]. Second, about 40% or more of patients with SDHB mutation have metastatic phenotype as the principal predictor of malignancy in PPGL patients [57,93,94]. Third, patients with genetic or syndromic PPGL have higher risks in development for PPGL complications compared to those with apparently sporadic disease [94,96]. Fourth, about 11% to 13% of patients with apparently sporadic PPGLs such as single benign PPGL without a family history have a mutation in susceptibility genes according to a meta-analysis [97]. Lastly, the identification of the genetic mutation in the proband may have implications for their family members related to the necessity of surveillance [98].
5.2. Genetic testing should be also considered for first-degree relatives of patients with hereditary PPGLs (B)
As mentioned in the previous section 5.1, PPGLs are the most heritable tumors with syndromic clinical features such as NF1, VHL, multiple endocrine neoplasia type 2 (MEN2), or paraganglioma syndrome (PGL). These pheochromocytoma-associated syndromes usually have a higher frequency of family history through the mostly autosomal dominant transmission [99]. For this reason, the Endocrine Society also recommends directly targeted germline mutation testing beyond sequential genetic testing in patients with PPGLs who have a positive family history or syndromic presentation [57]. Muth et al. [98] suggest that genetic testing should be applied to first-degree relatives in all hereditary PPGL and to second-degree relatives in case of SDHD, succinate dehydrogenase complex assembly factor 2 (SDHAF2)-related PPGLs as well as SDHB, SDHA, SDHC, FH, transmembrane protein 127 (TMEM127), and MAX mutation related to metastatic disease.
5.3. Validated targeted NGS is a preferred method for the genetic diagnosis of PPGLs (B)
Since 1990, germline mutation in a dozen PPGL susceptibility genes have been reported [57,95]. Moreover, as techniques for genetic testing have been advanced, higher mutation discovery rates and a better understanding of the clinical phenotypes of PPGLs have been achieved [93]. After the Endocrine Society proposed a clinical feature-driven diagnostic algorithm for priorities for specific genetic testing for PPGL, the advent of NGS methods offers a single assay for the screening of all PPGL susceptibility genes [100]. Therefore, decision algorithms for PPGL genetic testing has been changed from ‘from the patient to the candidate genes’ into ‘from the mutated genes to the affected patient [95]. Likewise, the implementation of NGS has been a paradigm shift in genetics research of PPGLs [92]. Accordingly, the NGS in PPGL (NGSnPPGL) study group recommended genetic testing should be performed in all patients with PPGLs independent of clear family history [92]. They preferred targeted NGS as a method for genetic diagnosis of PPGLs with blood (fresh collected within 7 days or frozen) or a frozen leukocyte pellet. The suggested targeted panels should include coding exons and intron boundaries of the targeted genes. As in conventional genetic testing, whenever possible, confirmation of the NGS-identified variant in a separate aliquot of the patient’s DNA is highly recommended by using an orthogonal method such as Sanger sequencing [92].
5.4. We recommend targeted NGS panels of gene sets based on the current level of evidence of their pathogenic driver status: 10 basic panel (FH, MAX, NF1, RET, SDHA, SDHB, SDHC, SDHD, TMEM127, VHL) and five extended panel (egl-9 family hypoxia inducible factor 1/prolyl hydroxylase domain 2 [EGLN1/PHD2], endothelial PAS domain-containing protein 1 [EPAS1], kinesin family member 1B [KIF1B], MET, SDHAF2) (C)
The NGSnPPGL study group proposed three sets of gene panels for the diagnosis of PPGLs; basic (evidence level 3 and 4 for germline mutations; including 10 genes such as FH, MAX, NF1, RET, SDHA, SDHB, SDHC, SDHD, TMEM127, and VHL), extended (evidence level 2 for germline mutations; including five more genes such as EGLN1/PHD2, EPAS HIF2A, KIF1B, MET, and SDHAF2), and comprehensive panels (all somatic mutations and all levels of evidence; extended genes added to 12 more genes including alpha thalassemia/mental retardation syndrome X-linked [ATRX], v-raf murine sarcoma viral oncogene homolog B1 [BRAF], cyclin dependent kinase inhibitor 2A [CDKN2A], EGLN2/PHD1, fibroblast growth factor receptor 1 [FGFR1], H3 histone, family 3A [H3F3A], Harvey rat sarcoma viral oncogene homolog [HRAS], isocitrate dehydrogenase 2 [IDH2], lysine [K]-specific methyltransferase 2D [KMT2D], malate dehydrogenase 2 [MDH2], MER proto-oncogene, tyrosine kinase [MERTK], and tumor protein p53 [TP53]) [92]. Herein, they modified ClinVar review status to adapt for their consensus statement on PPGLs driver genes. They used five levels (evidence level 0–5) based on numbers of applicable sources; level 0 with not applicable; level 1 with single source; level 2 as two or more sources without funcitonal validation; level 3 with two or more sources with some functional validation; and level 4 with established evidence from clinical, genetic, computational prediction as well as functional evidence and/or analysis of population frequency. A study about the diagnostic methods of PPGLs demonstrated that NGS assay significantly improved the performances of PPGL genetic testing compared with conventional methods with a higher rate of the identified mutation [101]. From that study, Ben Aim et al. [101] designed and suggested their custom multigene panel, ‘MASTER Plus SDHv2,’ including 17 PPGL genes; VHL, NF1, RET, SDHA, SDHB, SDHC, SDHD, SDHAF2, TMEM127, FH, MAX, EPAS1, EGLN1, EGLN2, MDH2, ATRX, and HRAS genes. Unfortunately, there are limited data on genetic studies of PPGLs in Korea [102]. Recently, a study with the largest number of Korean PPGL patients (n=161) reported that approximately 21% of sporadic PPGLs without any family history still harbored germline mutation of the PPGL-related genes [103]. This could be a convincing evidence for performing targeted NGS study in Korean patients with PPGLs like other ethnic groups. Another recent published data also reported genetic analysis and clinical characteristics of PPGLs in Korea [104]. In this study, among 57 patients with PPGLs, 28 different germline mutations were identified in 11 susceptibility genes (EPAS1, KIF1B, MAX, NF1, RET, SDHA, SDHB, SDHC, SDHD, TMEM127, and VHL). The prevalence of germline mutation was 32.6%, and the most frequently mutated genes were SDHB (n=11, 31.4%) followed by RET (n=8, 22.3%), VHL (n=6, 17.1%), NF1 (n=2, 5.7%), and EPAS1 (n=2, 5.7%) [104].
Taking all these considerations into account, we recommend two sets of gene panels (basic and extended) based on the current level of evidence of their pathogenic driver status, which adopted from the consensus statement of NGS based diagnostic testing of hereditary PPGLs by the NGSnPPGL study group (Table 6) [92]. When the primary mutation is not found in targeted NGS, comprehensive genetic studies such as whole-exome sequencing or whole-genome sequencing can be further considered to improve the understanding of the pathogenicity in any possible variants whenever possible [92]. A recent study performing WES in 36 patients with PPGL who had negative tests from previous Sanger sequencing or targeted gene panel revealed that two more likely pathogenic variants were additionally detected [105].

Recommended Targeted Next-Generation Sequencing Panels of Pheochromocytoma and Paraganglioma Based on Current Evidence
CONCLUSIONS
As a result of overarching research, PPGLs has been known as the representative hereditary tumor and newly classified as a malignant tumor. Also, great progress has been made regarding biochemical tests, functional imaging, and genetic testing. Accordingly, we can diagnose PPGLs more appropriately, detect multifocal or metastatic lesions, and predict prognosis better. We hope that this position statement provides the current knowledge and be helpful in clinical practice.
Acknowledgements
This research was supported by the Korean Endocrine Society. We would like to thank the Korean Endocrine Society, the Korean Surgical Society, the Korean Society of Nuclear Medicine, the Korean Society of Pathologists, and the Korean Society of Laboratory Medicine.
Notes
CONFLICTS OF INTEREST
No potential conflict of interest relevant to this article was reported.