Small Heterodimer Partner and Innate Immune Regulation
Article information

Abstract
The nuclear receptor superfamily consists of the steroid and non-steroid hormone receptors and the orphan nuclear receptors. Small heterodimer partner (SHP) is an orphan family nuclear receptor that plays an essential role in the regulation of glucose and cholesterol metabolism. Recent studies reported a previously unidentified role for SHP in the regulation of innate immunity and inflammation. The innate immune system has a critical function in the initial response against a variety of microbial and danger signals. Activation of the innate immune response results in the induction of inflammatory cytokines and chemokines to promote anti-microbial effects. An excessive or uncontrolled inflammatory response is potentially harmful to the host, and can cause tissue damage or pathological threat. Therefore, the innate immune response should be tightly regulated to enhance host defense while preventing unwanted immune pathologic responses. In this review, we discuss recent studies showing that SHP is involved in the negative regulation of toll-like receptor-induced and NLRP3 (NACHT, LRR and PYD domains-containing protein 3)-mediated inflammatory responses in innate immune cells. Understanding the function of SHP in innate immune cells will allow us to prevent or modulate acute and chronic inflammation processes in cases where dysregulated innate immune activation results in damage to normal tissues.
INTRODUCTION
Nuclear receptors (NRs) are transcriptional regulators of a diverse set of genes that play a central role in metabolic and endocrine homeostasis. NRs play key physiological roles in the regulation of gene networks that operate a variety of biological responses, including cellular growth and differentiation, development, cell cycle and proliferation, and metabolism [123]. Consequently, NRs and their modulating agents are emerging as promising therapeutic targets in various metabolic, reproductive, and proliferative disorders [3]. Most NRs are activated by endogenous ligands, but others have been classified as orphan NRs without known ligands [4]. Small heterodimer partner (SHP, NR0B2) is an atypical orphan NR, since the endogenous ligand has not been identified, with a unique structure and function that are distinct from conventional NRs [56].
In response to pathogenic or dangerous stimulation, the innate immune system is activated by triggering its numerous pattern-recognition receptors (PRRs) on innate cells, and induces pro-inflammatory and antimicrobial responses via intracellular signaling cascades [789]. It is clear that this PRR-mediated innate immune signaling is essential for the orchestration of the immediate host defense to infection and the subsequent activation of adaptive immunity [1011]. To avoid harmful immunopathological responses, the innate immune system is tightly regulated by a variety of molecules/pathways that control the magnitude of the inflammatory responses [1011].
Recently, we showed that SHP is an essential negative regulator of innate immune signaling and suppresses toll-like receptor (TLR) and NLRP3 (NACHT [NOD, NAIP, CIITA, HET-E, or TP1], leucine-rich repeat [LRR], and PYD domains-containing protein 3)-induced inflammasome activation in macrophages and in mice [121314]. The aim of this brief review was to discuss the unexpected role of SHP in macrophages in light of recent data on the molecular mechanisms by which SHP controls TLR-induced inflammation and NLRP3 inflammasome activation.
OVERVIEW OF SHP STRUCTURE AND FUNCTION
The SHP gene consists of two exons and a single intron located at human chromosome 1p36.1 subband [15]. The human SHP gene is expressed in major organs and tissues, including the liver, heart, pancreas, kidney, adrenal gland, spleen, stomach, and small intestine [151617]. The structure of SHP is relatively unique because it lacks a conventional DNA-binding domain, but has a putative ligand-binding domain, which makes SHP a member of the NR family [518]. Previous studies have shown that SHP exerts its transcriptional regulatory function through protein-protein interactions with a variety of NRs [61920]. For example, SHP interacts with several NR family members, including retinoid receptors, the thyroid hormone receptor, and the orphan receptor MB67 [5], etc. Through these interactions, SHP can affect diverse biological responses, including cholesterol, bile acid, triglyceride, and glucose homeostasis [62021].
Although SHP has no physiological ligands, numerous NRs and transcription factors (TFs) are able to target the SHP promoter and induce the expression of SHP. Earlier reports showed that steroidogenic factor 1 and the orphan fetoprotein transcription factor/liver receptor homologue 1 (LRH-1) can potently upregulate SHP promoter activity in tissues such as the adrenal glands and liver [22]. The farsenoid X receptor (FXR) was also shown to induce SHP transcription, which then inactivates LRH-1 to repress the expression of CYP7A1, the rate-limiting enzyme in the bile acid biosynthesis pathway; thus, resulting in the coordinated regulation of hepatic cholesterol metabolism [2324]. In addition, hepatocyte nuclear factor 4α (HNF4α) was also reported to regulate the transcription of FXR to inhibit the expression of SHP, which also heterodimerizes with HNF4α and inhibits its transcriptional activation [2526]. Moreover, other TFs, including c-Jun, basic helix-loop-helix TFs, and in particular the E2A proteins (E47, E12, and E2/5), function to activate SHP promoter activity [2728]. In addition, estrogen receptor-related receptor γ [16], liver X receptor α (LXRα) [29], peroxisome proliferator-activated receptor γ (PPAR-γ) [30], hepatocyte growth factor, and its family member macrophage-stimulating factor (MSP) were also involved in modulation of SHP promoter activity through upstream stimulatory factor 1 [31].
Several genetic variations, including single nucleotide polymorphisms and mutations in the human SHP gene, have been reported in relation to a variety of physiological responses [20]. A previous report showed SHP mutations in Japanese patients with obesity and diabetes, suggesting roles for SHP in insulin resistance and mild obesity [32]. In addition, SHP mutations identified in mildly obese subjects were found to be associated with an increased risk for type 2 diabetes later in life in Japanese patients [33]. However, the relationship between SHP genetic variations with diabetes or obesity has been controversial in European studies. Novel SHP missense mutations and polymorphisms were identified in subjects with severe obesity in the UK [34]. However, another study with 1,927 UK subjects showed no specific relationship between SHP genetic variation and type 2 diabetes, obesity, or birth weight [35]. In a cohort study of Danish people with early-onset obesity, several novel SHP variants were identified [36]. Therefore, these findings suggest the need for large-scale population studies to assess the relevant risk of human diseases in relation to SHP gene mutations and/or polymorphisms.
INNATE IMMUNITY: TLRs AND THE INFLAMMASOME
TLRs and nucleotide binding oligomerization domain (NOD)-like receptors (NLRs) are well-characterized membrane-bound and cytosolic PRRs, respectively [937]. They play important roles in innate recognition and inflammatory response elicitation against invading pathogens [9]. Both receptors have LRRs that are responsible for ligand interactions.
TLRs are transmembrane innate PRRs located at the cell surface and in the intracellular membrane. So far, 12 mouse and 10 human functional TLR members have been identified [38]. Each TLR can sense distinct pathogen-associated molecular patterns (PAMPs) from various pathogens, including lipoproteins (by TLR2/1 and TLR2/6), double-stranded RNA (by TLR3), and lipopolysaccharide (LPS; by TLR4) [38]. Other TLRs, including TLR3, 7, 8, and 9, are present on intracellular membranes and can recognize bacterial and viral nucleic acids [38]. TLR engagement by PAMP or damage-associated molecular patterns (DAMP) initiates overlapping and distinct signaling pathways in innate immune cells [938]. There are several principal adaptor proteins, including MyD88, MyD88 adapter-like (MAL), Toll/IL-1 receptor domain-containing adaptor inducing β (TRIF), and Trif-related adaptor molecule (TRAM), that contain Toll/interleukin-1 receptor homology (TIR) domains and mediate the intracellular signaling triggered by TLRs [3940]. Most TLRs, except for TLR3, are able to transmit signals through the adaptor protein MyD88, whereas TLR3 and TLR4 signal through TRIF [383940].
Twenty-two NLR family members that contain a C-terminal LRR domain, a central NACHT domain, and an N-terminal effector motif have been described [41]. NOD1 and NOD2 detect diaminopimelatic acid-containing muropeptide, primarily found in gram-negative bacteria, and muramyl dipeptide (MDP) moieties, usually found in all bacterial peptidoglycans, respectively, to initiate an innate inflammatory response and host defense pathways [4243444546]. Pyrin domain-containing proteins (NLRPs) are another group of NLRs; of these NLRs, NLRP3 is the best characterized, and activates inflammasome assembly [46].
Inflammasomes are an intracellular molecular platform of multiprotein complexes that activate caspase-1, resulting in the maturation of pro-interleukin-1β (pro-IL-1β) to IL-1β and its subsequent release [47484950]. Numerous PAMP and DAMP signals can activate the NLRP3 inflammasome complex, including adenosine triphosphate, MDP, uric acid crystals, cholesterol crystals, silica, and specific pathogenic infections [4649505152]. Indeed, NLRP3 inflammasome activation is mediated through common general molecular pathways such as increased potassium efflux, lysosomal damage, or mitochondrial reactive oxygen species (ROS) generation [46535455]. Currently, a two-step process is generally accepted for NLRP3 inflammasome activation. The first "priming" step involves nuclear factor κB (NF-κB)-dependent production of inactive precursors pro-IL-1β and pro-IL-18 and inflammasome components, including NLRP3 [56]. Following activation, NLRP3 can assemble the caspase-1-dependent inflammasome by recruiting an adaptor protein apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC), also known as TMS1 or PYCARD, and procaspase-1 [465354]. A second signal is required for NLRP3 inflammasome assembly by the recruitment of ASC and procaspase-1 to activate caspase-1 and to release mature cytokines IL-1β and IL-18 [465354].
Previous reports have shown that NLRP3 mutations are involved in an autoinflammatory disease, cryopyrin-associated periodic syndrome, which manifests as systemic inflammation and serious complications [57]. Therapeutic intervention using IL-1β neutralization is highly beneficial in these patients [57]. Accumulating evidence also suggests that NLRP3 is involved in inflammatory bowel disease, cardiovascular disease, and rheumatoid arthritis [58]. In addition, numerous studies showed that NLRP3 inflammasome activation is important for the pathogenesis of metabolic syndromes, such as insulin resistance and obesity [59]. Moreover, dysregulated activation of the NLRP3 inflammasome seems to be implicated in the pathogenesis of other chronic inflammatory diseases, including Alzheimer disease [60], atherosclerosis [61], and age-related macular degeneration [6263]. Therefore, recent efforts to modulate NLRP3 inflammasome complex activation may create new opportunities to treat or prevent numerous chronic inflammatory and metabolic diseases.
SHP REGULATION OF INNATE IMMUNITY AND INFLAMMATION
Since innate immune activation should be tightly regulated to prevent unwanted host damage, numerous studies have identified the roles of negative regulators in TLR signaling and NLRP3 inflammasome activation [6465]. Recently, we described a previously unknown role for SHP in the negative regulation of TLR signaling [1314]. During TLR engagement, proinflammatory cytokines are produced by a series of intracellular mediators including interleukin-1 receptor-associated kinase 1, TNF receptor-associated factor 6 (TRAF6), and NF-κB in macrophages. SHP is essential for the regulation of TLR-mediated transactivation of canonical NF-κB and Lys63-linked polyubiquitination of TRAF6. In unstimulated cells, SHP is associated with the NF-κB p65 subunit in the cytoplasm and inhibits the nuclear translocation of NF-κB p65, resulting in the attenuation of proinflammatory transcriptional activity. TLR4 stimulation leads to rapid dissociation of SHP from the NF-κB p65 subunit, and SHP then interacts with the TRAF6 really interesting new gene (RING) domain that plays a crucial role in TRAF6 ubiquitination and activation [13].
Our previous studies demonstrated the possibility of SHP-inducing drugs as therapeutic candidates for systemic inflammatory diseases. Although SHP-specific ligands have not yet been identified, treatment with various AMP-activated protein kinase (AMPK)-activating drugs, such as MSP and fenofibrates, induce the expression of SHP in hepatocytes [3166] and macrophages [1314]. In addition, we demonstrated that liver kinase B1-dependent AMPK activation is required for the expression of SHP in macrophages treated with MSP and fenofibrate. Several studies have shown that MSP plays an inhibitory role in the LPS-mediated macrophage production of nitric oxide [6768], and the mechanisms by which MSP proteins are involved in TLR signaling involve the attenuation of serine phosphorylation of NF-κB p65 and NF-κB-dependent activation of IκBζ [69]. Our study further showed that MSP has a regulatory function in TLR4-mediated inflammatory responses through prevention of TRAF6 polyubiquitination. Moreover, fenofibrate is a well-known ligand of PPARα that has an anti-inflammatory effect in acute respiratory distress syndrome [70] and retinal hyperinflammation of type 1 diabetes-induced retinopathy [71]. However, fenofibrate improves survival in mice and inhibits the production of proinflammatory cytokines and mitochondrial ROS through PPARα-independent AMPK-SHP-uncoupling protein 2 (UCP2) regulatory cascades in TLR4-mediated systemic inflammatory responses. These data strongly suggest that the development and discovery of potential candidates involved in SHP induction and activity modulation may partially control systemic inflammatory diseases.
Previous studies suggested that the inflammasome plays a crucial role in host immune defense during infection with a variety of pathogens such as microbes, virus, and parasites [727374]. However, uncontrolled activation of inflammasomes leads to autoinflammatory diseases that are associated with the production of inflammatory cytokines such as IL-1β [7576]. We recently identified an unrecognized role for SHP in the regulation of the NLRP3 inflammasome [12]. SHP deficiency results in enhanced activation of the NLRP3 inflammasome, while abundant SHP protein produced by SHP-inducing agents, including MSP and fenofibrate, competitively inhibits the physical interaction between NLRP3 and ASC, and then intercepts the assembly of the NLRP3 inflammasome complex, which eventually leads to down-regulation of IL-1β and IL-18 production in NLRP3-activated macrophages. Moreover, SHP is translocated to the mitochondria with the NLRP3-ASC complex after inflammasome activation and then acts to regulate mitochondrial homeostasis through the recovery of inflammasome-induced mitochondrial ROS generation and damage. These findings demonstrate that SHP plays a fine-tuning role to prevent an excessive inflammatory response (Fig. 1).
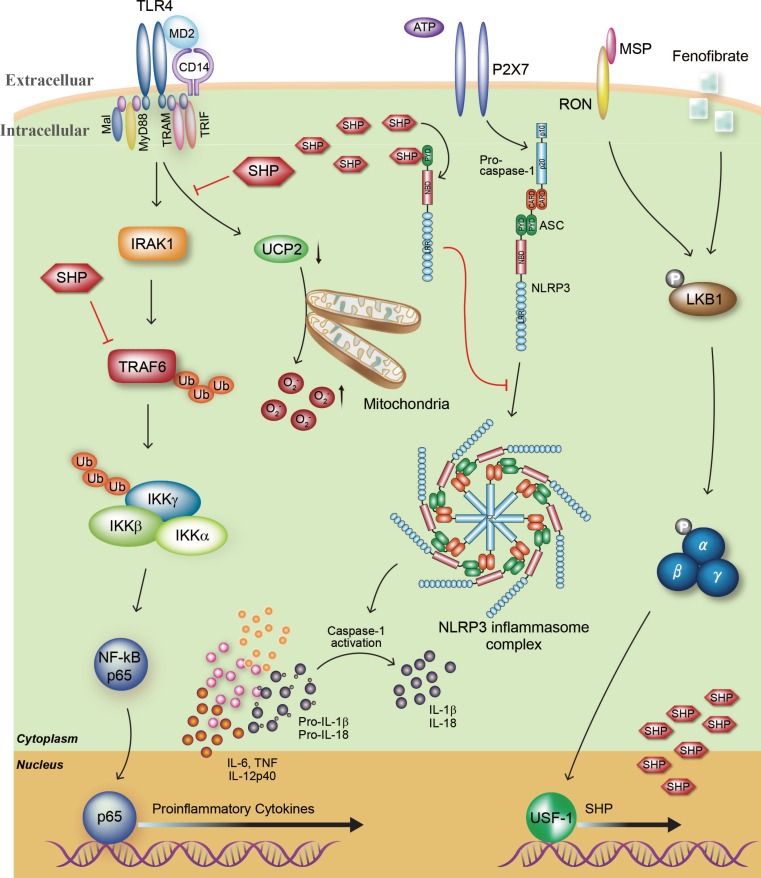
Roles of the orphan nuclear receptor small heterodimer partner (SHP) in the toll-like receptor 4 (TLR4)-mediated inflammatory response and NLRP3 (NACHT, LRR and PYD domains-containing protein 3) inflammasome activity. The orphan nuclear receptor SHP plays an important role in the negative regulation of lipopolysaccharide (LPS)-meditated inflammation and NLRP3 inflammasome activation. First, TLR4 engagement strongly acts to generate proinflammatory cytokines including tumor necrosis factor α (TNF-α), interleukin 6 (IL-6), IL-1β, and IL-12p40 through TNF receptor-associated factor 6 (TRAF6) ubiquitination and nuclear factor κB (NF-κB) p65 nuclear translocation. Moreover, the generation of mitochondrial reactive oxygen species (ROS) in macrophages is enhanced during LPS stimulation, which is closely associated with activation of the inflammatory response. The mitochondrial anion carrier protein uncoupling protein 2 (UCP2) is an essential component of mitochondrial ROS and inflammation modulation. Our previous studies indicated that endogenous SHP inhibits the TLR4-induced upregulation of proinflammatory responses through the modulation of TRAF6 polyubiquitination and UCP2 expression. Second, inflammasome activation results in the maturation of pro-IL-1β and pro-IL-18 and the secretion of these mature cytokines into the extracellular environment. SHP deficiency causes enhanced secretion of mature IL-1β and IL-18. On the other hand, overexpression of SHP effectively attenuates NLRP3 activation and secretion of IL-1β and IL-18 through direct interaction with NLRP3 protein. Third, treatment with fenofibrate or macrophage-stimulating factor (MSP) activates SHP gene expression through liver kinase B1 (LKB1)-dependent AMP-activated protein kinase activation and recruitment of upstream stimulatory factor-1 (USF1) to the human SHP promoter. In addition, in vivo administration of SHP-inducing drugs effectively protects against LPS-induced lethal shock and folic acid-induced acute tubular necrosis. MD2, lymphocyte antigen 96; TRAM, Trif-related adaptor molecule; TRIF, Toll/IL-1 receptor domain-containing adaptor inducing β; RON, recepteur d'origine nantais; NBD, nucleotide-binding domain; CARD, caspase activation and recruitment domains; PYD, pyrin domain; ASC, adaptor protein apoptosis-associated speck-like protein containing a caspase recruitment domain; IRAK1, interleukin-1 receptor-associated kinase 1; IKK, IκB kinase.
CONCLUSIONS
Accumulating evidence strongly suggests that NRs are crucial regulators of the physiological responses related to the progression of various diseases, including inflammatory and metabolic diseases. Because an uncontrolled or excessive inflammatory response generally leads to venomous pathogenesis with a high mortality rate, many lines of research have focused on the negative regulation of this process, and numerous negative regulators have been identified as a result. Several NRs, such as the glucocorticoid receptor, pregnane X receptor, PPAR-γ, and LXRs, are involved in the regulation of TLR signaling through post-translational or transcriptional repression. In this issue, we focus on the regulatory roles of orphan NR SHP during activation of the TLR-mediated inflammatory response and NLRP3 inflammasome activity. We previously demonstrated that the regulation of TLR signaling and NLRP3 inflammasome activity by SHP and gene expression of SHP are closely related, which suggests functional autoregulation of SHP during the innate immune response. In brief, SHP interacts with cytosolic p65 in resting cells, whereas TLR stimulation leads to rapid dissociation between SHP and p65. The cytosolic SHP protein then interacts with TRAF6, thereby completing the polyubiquitination of TRAF6. Moreover, SHP deficiency results in enhanced generation of mitochondrial ROS that is related to the decrease in mitochondrial anion carrier protein UCP2 protein expression. During activation of the NLRP3 inflammasome, SHP temporarily interacts with endogenous NLRP3 in the perinuclear region, translocates to the mitochondria to maintain mitochondrial homeostasis, and then attenuates caspase-1 activation and IL-1β and IL-18 cytokine maturation. Taken together, these studies indicate that SHP is an intrinsic negative regulator of NLRP3 inflammasome and TLR-mediated inflammatory responses. Moreover, our results strongly suggest that therapeutic or protective drugs, such as MSP and fenofibrate, which modulate SHP induction and activity, partially control inflammatory diseases including endotoxic shock and acute tubular necrosis. Understanding the roles of SHP and various NRs in animals and humans will facilitate the development of novel therapeutics to treat inflammatory diseases.
ACKNOWLEDGMENTS
We are indebted to current and past members of our laboratory for discussions and investigations that contributed to this article. This work was supported by research fund of Chungnam National University and the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP; No. NRF-2015M3C9A2054326). I apologize to colleagues whose work and publications could not be referenced owing to space constraints.
Notes
CONFLICTS OF INTEREST: No potential conflict of interest relevant to this article was reported.