Partial Deletion of Perk Improved High-Fat Diet-Induced Glucose Intolerance in Mice
Article information

Abstract
Although pancreatic endoplasmic reticulum kinase (PERK) is indispensable to beta cells, low-dose PERK inhibitor improved glucose- stimulated insulin secretion (GSIS) and hyperglycemia in diabetic mice. Current study examined if partial deletion of Perk (Perk+/-) recapitulated the effects of PERK inhibitor, on the contrary to the complete deletion. Perk+/- mice and wild-type controls were fed with a high-fat diet (HFD) for 23 weeks. Glucose tolerance was evaluated along with serum insulin levels and islet morphology. Perk+/- mice on normal chow were comparable to wild-type mice in various metabolic features. HFD-induced obesity was not influenced by Perk reduction; however, HFD-induced glucose intolerance was significantly improved since 15-week HFD. HFD-induced compromises in GSIS were relieved by Perk reduction, accompanied by reductions in phosphorylated PERK and activating transcription factor 4 (ATF4) in the islets. Meanwhile, HFD-induced islet expansion was not significantly affected. In summary, partial deletion of Perk improved glucose tolerance and GSIS impaired by diet-induced obesity, without changes in body weights or islet mass.
INTRODUCTION
Pancreatic endoplasmic reticulum kinase (PERK) is a protein in the endoplasmic reticulum (ER) and plays critical roles in pancreatic beta cells. Genetic compromise of PERK induced pancreatic atrophy and insulin insufficiency, along with early diabetes [1,2].
In contrast to ablation of Perk expression, chronic treatment of PERK inhibitors GSK2606414 and GSK2656157 at low doses rather improved glucose-stimulated insulin secretion (GSIS) in mouse and human islets [3], improving hyperglycemia in a mouse model of type 2 diabetes [4]. Meanwhile, the PERK inhibitors may have unexpected targets besides PERK: they were proposed to repress receptor-interacting serine/threonine kinase 1 (RIPK1) kinase activity, independently on PERK [5]. Therefore, it is unclear if the anti-diabetic effects of PERK inhibitors were mediated by attenuation of PERK or RIPK1 or both.
Therefore, we examined if partial reduction of Perk leading to attenuation of PERK activity would recapitulate effects of synthetic PERK inhibitors in mice.
METHODS
Animal study
We used male Perk+/-mice (from Dr. John C. Bell, University of Ottawa) and the wild-type littermates, with mixed background of C57BL/6 and BALB/c. Genotyping by polymerase chain reaction (PCR) demonstrated the wild-type allele and mutant allele as bands of 231 and 302 bp, respectively (primer sets in Supplemental Table S1) [6]. High-fat diet (HFD) was fed for 23 weeks from 5 weeks of age. Insulin levels were measured using an enzyme-linked immunosorbent assay (ELISA) kit (80-INSMS-E01, ALPCO, Salem, NH, USA), from serum obtained before and after intraperitoneal glucose injection (1 g/kg weight). All the animal studies were performed in accordance with the Institutional Animal Care and Use Committee (SNU-190808-1, 22-0281-S1A1).
Islet expression of unfolded protein response markers
Mouse islets were isolated as previously, and total RNA or protein were extracted for reverse transcription PCR and Western blotting (Supplemental Tables S2, S3) [4].
Islet morphology
After 23-week HFD, pancreas was harvested and embedded in paraffin. Microscopic examination was conducted after hematoxylin and eosin staining and immunohistochemical staining (Supplemental Table S3) using Aperio Digital Pathology and ImageScope (Leica Biosystems, Wetzlar, Germany).
Statistical analysis
Data are expressed as mean±standard error of the mean. Statistical analyses were executed using Prism (GraphPad, San Diego, CA, USA). P values <0.05 were considered statistically significant.
RESULTS
Characteristics of Perk+/- mice
Reduction of Perk expression was confirmed by RNA and protein levels in islets (Fig. 1A). Body weights, blood glucose levels, glucose tolerance, insulin tolerance, and GSIS were comparable between the genotypes during age of 9 to 18 weeks (Fig. 1B-F). Reduction of Perk did not affect pancreatic weights and islet morphology (Fig. 1G, H).

Metabolic phenotypes and islet morphology of pancreatic endoplasmic reticulum kinase (Perk)+/- mice. Adult male Perk+/- mice and male wild-type littermates were compared after confirmation of the genotypes and Perk expression. Intraperitoneal glucose tolerance test (1 g/kg weight) and insulin tolerance test (regular insulin 0.5 U/kg weight) were performed after overnight fasting at 20 weeks old. (A) Pictures of reverse transcription-polymerase chain reaction (left) and Western blotting (right) using isolated islets. (B) Body weights. (C) Fed blood glucose levels. (D) Glucose tolerance test. (E) Insulin tolerance test. (F) Serum insulin levels before and after glucose loading. (G) Pancreas weights. (H) Representative pancreatic sections of hematoxylin and eosin staining. For (B-E), two-way repeated-measures analysis of variance was used. Paired t test for (F) and Student’s t test for (G) were applied. No statistical differences were found. Animal numbers=10 to 16 for each genotype. Gapdh, glyceraldehyde-3-phosphate dehydrogenase.
Changes in glucose levels by HFD
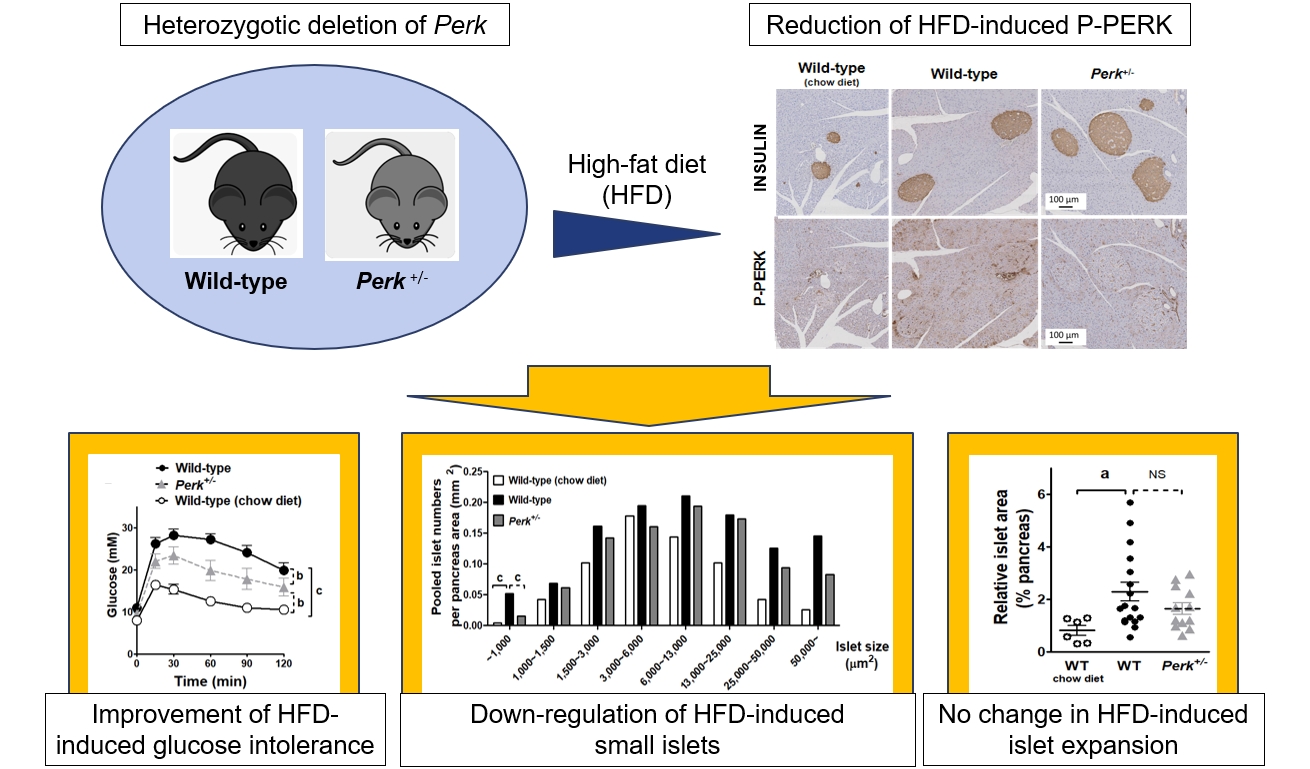
HFD-induced weight gain without difference between the genotypes (Fig. 2A). HFD-induced hyperglycemia in the wild-type mice (P<0.001) (Fig. 2B). Perk+/- mice showed significantly lower glucose levels compared to the wild-type since 17 weeks of HFD (9.2 mM vs. 11.5 mM in average, P<0.05). HFD-induced glucose intolerance was not affected by Perk reduction after 5-week HFD (Fig. 2C); however, it was significantly improved after 15-week HFD (by 46% of HFD-induced increment in area under the curve, P<0.01) (Fig. 2D).
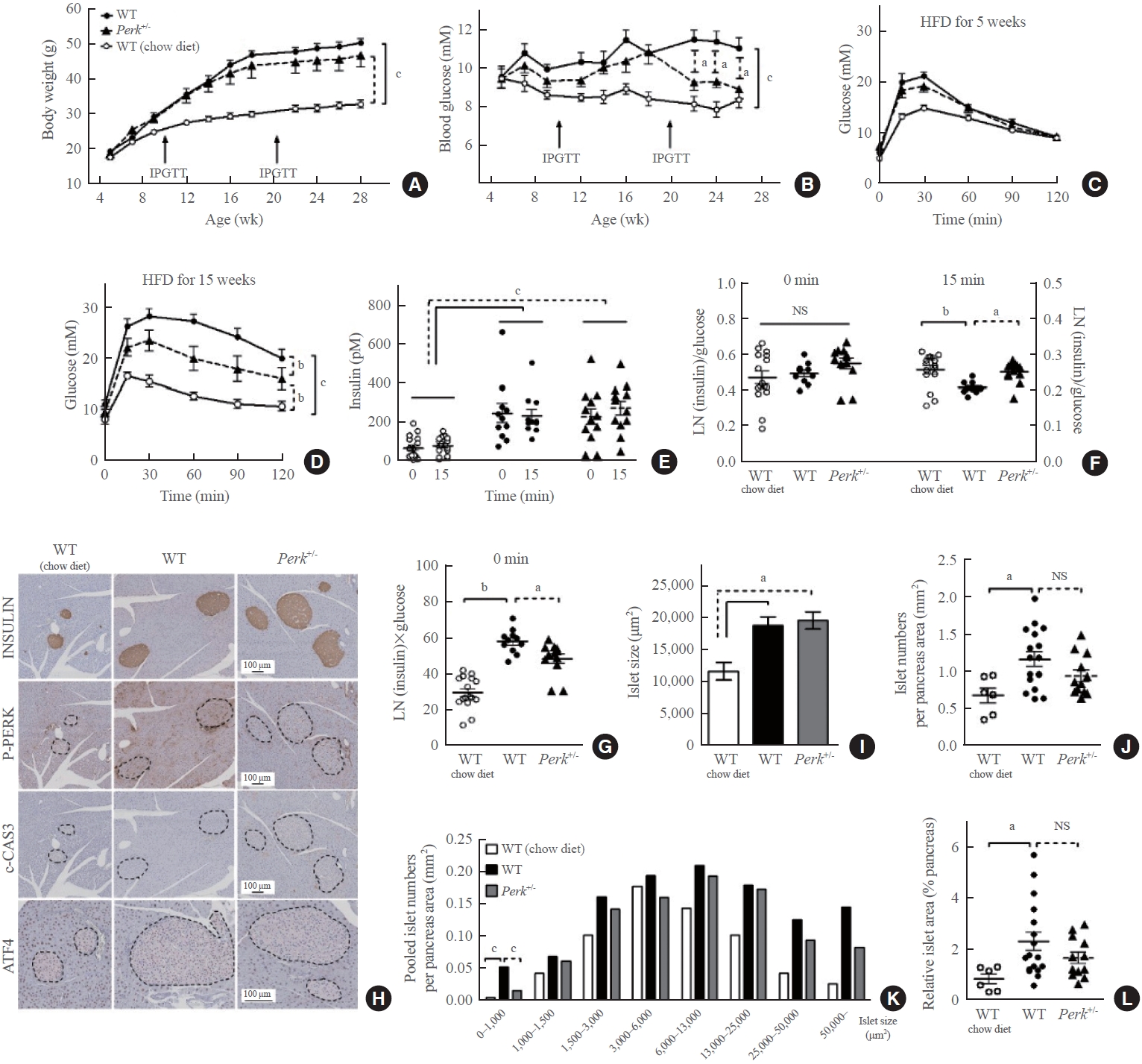
Changes in the metabolic phenotypes and islet morphology by high-fat diet (HFD) in pancreatic endoplasmic reticulum kinase (Perk)+/- mice. Mice of either genotype were given a HFD at age 5 weeks for 23 weeks, and compared to wild-type (WT) mice on chow-diet. Body weights (A) and random blood glucose levels (B) were monitored. After 5 and 15 weeks of HFD, intraperitoneal glucose tolerance tests were performed (1 g/kg weight) (C, D). Serum insulin levels were measured before and 15 minutes after the glucose loading after 15-week HFD (E), and were subject to assess insulin secretion by adjustment with glucose levels (F). Insulin resistance was estimated by multiplying fasting insulin and glucose levels (G). Representative pictures of immunohistochemical stains in the pancreas (islets marked by broken lines) extracted after 23-week HFD or normal chow (H), pooled analysis for average islet sizes (I), islet numbers on each section adjusted by the pancreas area (J), islet size distribution after pooling the islets in each group (K), and relative islet area adjusted by the pancreas area (L). For (A-D), two-way repeated-measures analysis of variance (ANOVA) and Bonferroni posttests were performed. For the others, one-way ANOVA and Bonferroni posttests were applied, except for (K) where qui-square test was applied. Animal numbers, 7 to 17 for each group; pancreas area, 37.7±2.5 mm2/section; islet numbers, median 35/section from a mouse. IPGTT, intraperitoneal glucose tolerance test; NS, no significant difference; LN, logarithmus naturalis; P-PERK, phosphorylated PERK; c-CAS3, cleaved caspase-3; ATF4, activating transcription factor 4. aP<0.05; bP<0.01; cP<0.001 between the indications.
Changes in insulin levels by HFD
The HFD-induced hyperinsulinemia without significant difference between the genotypes (Fig. 2E). Insulin stain intensities on the pancreas sections were also comparable (Fig. 2H). When the plasma insulin levels were adjusted by the corresponding glucose levels, either HFD or Perk reduction did not influence insulin-to-glucose ratio at fasting, while HFD suppressed the ratio after glucose loading, by 20% (P<0.01) (Fig. 2F). It was recovered by Perk reduction (P<0.05). Fasting insulin multiplied by glucose levels suggesting insulin resistance significantly increased by HFD in the control mice by 2-fold, and it was slightly lower in Perk+/- mice than in the wild-type (P<0.05) (Fig. 2G).
Changes in unfolded protein response markers in the islets by HFD
According to the immunohistochemical staining, HFD-induced phosphorylated PERK (P-PERK) was significantly down-regulated in the Perk+/- mice. However, cleaved caspase-3 was not induced by HFD in the islets of either genotype (Fig. 2H), indicating that adapted unfolded protein response had prevented ER stress. Nuclear activating transcription factor 4 (ATF4) stain in the islets seemed slightly down-regulated in the Perk+/ mice compared to the wild-type. Reductions in the P-PERK along with phosphorylated eukaryotic translation initiation factor 2A (P-EIF2A) and ATF4, with no significant changes in the final index of ER stress C/EBP homologous protein (CHOP), were reproduced in the Perk+/ islets exposed to chronic lipotoxicity in vitro (Supplemental Fig. S1).
Changes in islet morphology by HFD
HFD increased average islet sizes in both the genotypes by 65% (Fig. 2I). HFD increased islet numbers in the wild-type (0.64 vs. 1.11/mm2 pancreas, P<0.05) (Fig. 2J). Despite a tendency to decrease in the numbers by Perk+/- reduction (0.92/mm2), there was no statistical difference between the genotypes. Next, islet numbers according to islet sizes revealed that frequency of small islets (<1,000 μm2) was different from that of the other sizes: more islets in the HFD-fed wild-type, which was significantly inhibited in the Perk+/- mice (P<0.001) (Fig. 2K). Finally, islet area relative to the pancreas area increased in wild-type mice by HFD by 2-fold with an insignificant tendency to decrease by Perk reduction (Fig. 2L).
DISCUSSION
In this study, reduction of Perk relieved HFD-induced P-PERK in the islets (Fig. 2H) and impairment of GSIS in mice (Fig. 2F), just like administration of low-dose PERK inhibitor [4]. These findings were accompanied by improvement of glucose intolerance, only after prolonged HFD (Fig. 2B-D), suggesting beta cell exhaustion which could not be reproduced in vitro (Supplemental Fig. S2) [7].
Initially, Perk+/- mice were reported slightly glucose intolerant [6]. However, Wang et al. [8] found that Perk+/- mice exhibited hyperinsulinemia during neonatal development, causing transient hypoglycemia. They observed that Perk reduction in pancreas, but not in the liver, decreased blood glucose levels. Different genetic backgrounds and ages might explain the conflicting results. In a pathologic environment, Gupta et al. [9] observed that a reduction of Perk dosage in Akita mice with insulin deficient diabetes increased pancreatic insulin contents and improved hyperglycemia, compatible with our current study.
Expansion of beta cell mass is caused by altered balance between beta cell death and generation [10]. Current study reproduced HFD-induced increases in islet sizes and numbers [11]. Because PERK is critical to beta cells generation [2], the suppression of HFD-induced small islets in Perk+/- mice (Fig. 2K) might reflect inhibition of islet neogenesis due to low PERK activity, and/or secondary to less severe insulin resistance (Fig. 2G). Down-regulation of ATF4 (Fig. 2H, Supplemental Fig. S1) may have a role in the changes in the function and generation of Perk+/- islet. Intriguingly, partial Perk reduction has been reported to increase mature beta cell mass under physiologic condition [8]. PERK inhibitor at low doses did not influence beta cell apoptosis and mass in diabetic mice [4], suggesting that beta cell generation did not change significantly. Further studies are required to answer these complicated questions.
We also observed that HFD-induced insulin resistance partially improved in the Perk+/- mice (Fig. 2G). P-PERK significantly increased in the liver of obese mice [12], which may decrease insulin sensitivity through forkhead box protein O1 (FOXO1) [13]. Blocking of PERK activity has attenuated fatty acid-induced insulin resistance in hepatocytes [14]. Therefore, the improved insulin resistance might have contributed to the improved glucose intolerance in the Perk+/- mice.
There are several weak points in this study. Glucose loading did not induce significant insulin levels (Fig. 2E), which reflects inappropriate dose of glucose and/or the measurement time [15]. However, GSIS calculated by insulin-to-glucose ratio could support our hypothesis, instead. Another point is that islet cell composition was not evaluated. Critically, this study cannot tell contribution of RIPK1 in the effects of PERK inhibitors. It requires animal models with RIPK1-insufficient islets. Indeed, inhibition of RIPK1 is known to enhance islet function [16]; therefore, PERK inhibitors might have anti-diabetic effects through attenuation of both PERK and RIPK1.
In conclusion, attenuation of PERK activity by genetic suppression improved GSIS and hyperglycemia in a mouse model of obesity-induced diabetes, supporting PERK as a new target for diabetes therapy.
Supplementary Material
Supplemental Table S1.
Primer Sequences for Genotyping Polymerase Chain Reaction
Supplemental Table S2.
Primer Sequences for Reverse Transcription-Polymerase Chain Reaction
Supplemental Table S3.
Antibodies for Immunohistochemical Staining and Western Blotting
Supplemental Fig. S1.
Western blots of pancreatic endoplasmic reticulum kinase (PERK) and unfolded protein response markers in mouse islets exposed to chronic lipotoxicity in vitro. Mouse islets were isolated from each genotype, and exposed to 0.5-mM palmitate (PA) (conjugated with bovine serum albumin free from fatty acid [3:1 molar ratio]) for 48 hours. We extracted total proteins from the islets using radioimmunoprecipitation assay (RIPA) buffer (BRI-9010, T&I) mixed with a protease inhibitor cocktail and phenylmethylsulfonyl fluoride. We boiled the protein samples in sodium dodecyl-sulfate (SDS) sample buffer, separated them by SDS-polyacrylamide gel electrophoresis, and then transferred them to nitrocellulose membranes for immunoblotting. We detected the blots using the ECL Western Blotting Substrate (Thermo, #NCI4080KR). Primary antibodies used are presented in the Supplemental Table S3. (A) Representative Western blots, and (B) the quantitated protein levels relative to TUBULIN. P-PERK, phosphorylated PERK; P-EIF2A, phosphorylated EIF2A; ATF4, activating transcription factor 4; CHOP, C/EBP homologous protein; NS, no significant; WT, wild-type. aP<0.05; bP<0.01 in the one-way analysis of variance (ANOVA) and Dunnett’s multiple comparisons test.
Supplemental Fig. S2.
Glucose-stimulated insulin secretion (GSIS) from mouse islets and the islet insulin contents exposed to in vitro lipotoxicity. Mouse islets were isolated from each genotype, and exposed to 0.5-mM palmitate (conjugated with bovine serum albumin free from fatty acid [3:1 molar ratio]) for 24 hours. After starvation and incubation with low glucose (2.8 mM) and high glucose (17.5 mM), we collected the supernatants and assayed the insulin using an enzyme-linked immunosorbent assay (ELISA) kit (ALPCO). Then we collected the islets, extracted insulin using HCl, and assayed it using the same ELISA kit. (A) GSIS from mouse islets and (B) islet insulin contents. For (A), two-way repeated-measures analysis of variance (ANOVA) was used. For (B), one-way ANOVA was used. NS, no significant difference; LG, low glucose; HG, high glucose; WT, wild-type; Perk, pancreatic endoplasmic reticulum kinase. aP<0.05 between the indication.
Notes
CONFLICTS OF INTEREST
No potential conflict of interest relevant to this article was reported.
AUTHOR CONTRIBUTIONS
Conception or design: M.J.K., H.S.J. Acquisition, analysis, or interpretation of data: J.L., M.J.K., S.M., J.Y.L. Drafting the work or revising: J.L., K.S.P., H.S.J. Final approval of the manuscript: J.L., M.J.K., S.M., J.Y.L., K.S.P., H.S.J.
Acknowledgements
This study was supported by NRF (grant number 2022R1A2C 2004570) funded by the Ministry of Science and ICT, Republic of Korea and Seoul National University Hospital Research Fund #0320230310. The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
We are grateful to Drs. John C. Bell (University of Ottawa, Canada) and Seung-Yong Seong (Seoul National University, Korea) for generous provision of Perk+/- mice, and to Dr. Young Joo Park (Seoul National University, Korea) for technical assistance.