The Role of Macrophage Lipophagy in Reverse Cholesterol Transport
Article information

Abstract
Macrophage cholesterol efflux is a central step in reverse cholesterol transport, which helps to maintain cholesterol homeostasis and to reduce atherosclerosis. Lipophagy has recently been identified as a new step in cholesterol ester hydrolysis that regulates cholesterol efflux, since it mobilizes cholesterol from lipid droplets of macrophages via autophagy and lysosomes. In this review, we briefly discuss recent advances regarding the mechanisms of the cholesterol efflux pathway in macrophage foam cells, and present lipophagy as a therapeutic target in the treatment of atherosclerosis.
INTRODUCTION
Atherosclerosis is a chronic inflammatory disease characterized by the development of lipid-rich plaques that inhibit arterial blood flow [12]. Animal experiments and human specimen investigations have established that hypercholesterolemia promotes the inflammatory processes leading to atherosclerosis. Hypercholesterolemia induces the accumulation of apolipoprotein B (apoB)-rich lipoprotein, the main protein in atherogenic lipoprotein particles such as low density lipoprotein (LDL), very low density lipoprotein (VLDL), and lipoprotein(a), in the intima under the endothelial cell layer, leading to the recruitment of monocytes and initiation of the immune response. These monocyte-derived macrophages play an important role throughout the entire process of atherogenesis. Interestingly, recent studies have revealed that macrophage lipophagy has a novel function in contributing to the development of vascular disease. In this review, we discuss the role of macrophages in cholesterol metabolism in reverse cholesterol transport (RCT) and the contribution of macrophage lipophagy to atherosclerosis (Fig. 1).
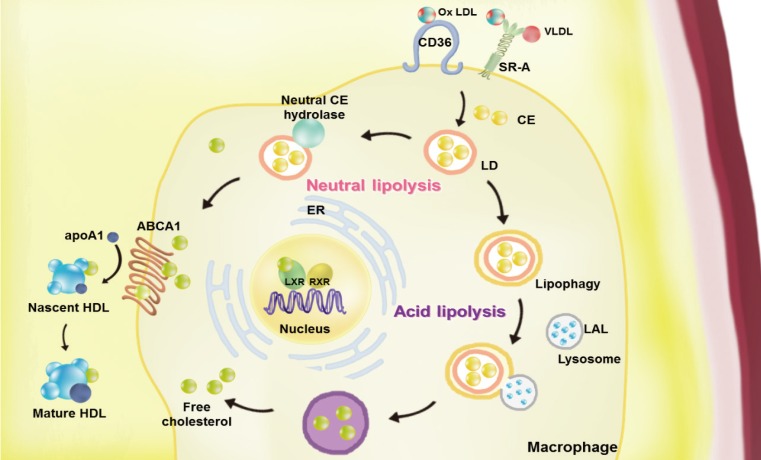
Overview of the pathways of macrophage lipoprotein uptake and efflux. Macrophages uptake very low density lipoprotein (VLDL) and modified low density lipoprotein (LDL), such as oxidized (Ox) LDL via scavenger receptors (SRs, including by SR-A1 and cluster of differentiation 36 [CD36]). The internalized LDL is esterified by acetyl-coenzyme A acetyltransferases (ACAT1) and stored in lipid droplets (LDs). Neutral and acid lipolysis contribute to the release of cholesteryl ester (CE) for efflux in LDs via neutral CE hydrolase or lipophagy through lysosomal acid lipase (LAL). The cellular free cholesterol activates the liver X receptor (LXR)-retinoid X receptor (RXR) heterodimeric transcription factor that upregulates expression of ATP-binding cassette subfamily A member 1 (ABCA1). This transporter mediates the free cholesterol efflux from macrophages, with lipid-poor apolipoprotein A1 (apoA1) used as an acceptor. By reducing the accumulation of cholesterol in the wall of arteries via macrophage cholesterol efflux, reverse cholesterol transport may the prevent development of atherosclerosis. ER, endoplasmic reticulum; HDL, high density lipoprotein.
MACROPHAGES IN REVERSE CHOLESTEROL TRANSPORT
RCT is a process through which excess cholesterol from peripheral cells and tissues returns to the liver for excretion, and plays an important role in reducing atherosclerosis. Macrophage cholesterol efflux is the first step of RCT that occurs in atherosclerotic vessel wall by macrophage-specific. In the early stages of atherogenesis, apoB-lipoproteins that have entered the intima are modified by processes such as oxidation and hydrolysis. These modifications lead to lipoprotein aggregation and further promote lipoprotein retention in the vessel wall [3]. The inflammatory signals originating from modified lipoproteins trigger endothelial activation and monocyte recruitment into the intima, and cause monocytes to differentiate into macrophages. Macrophages uptake modified lipoproteins within the cytoplasm through scavenger receptors (SRs), resulting in foam cell formation. Foam cell formation is the initial and key event of atherosclerosis.
Macrophage scavenger receptors
Although macrophages can clear modified LDL within intima through the low density lipoprotein receptor (LDLR), because the expression of LDLR is reduced in the early stage of foam cell formation by a decrease in sterol regulatory element-binding protein (SREBP)-1c due to increased cellular cholesterol levels [4], macrophages use another type of membrane receptor for apoB-lipoprotein removal. SRs are a diverse range of transmembrane proteins that internalize modified LDL and lipoprotein-base ligands. SRs have been divided into eight subclasses that share the defining feature of being able to bind various forms of modified LDL [5]; additionally, they can perform a wide variety of other functions, such as phagocytosis, antigen presentation, and elimination of apoptotic cells, depending on what they bind to. Nevertheless, the first and most important receptors that are responsible for modified LDL uptake in intimal macrophages are scavenger receptors type 1 (SR-A1) and cluster of differentiation 36 (CD36) [6].
SR-A and CD36 are responsible for 75% to 90% of modified LDL degradation, and macrophages harvested from SR-A/CD36 double-null mice show an abnormal accumulation of cholesteryl esters (CEs) derived from modified LDL [7]. It has been evidently revealed in vitro that SR-A1 and CD36 play a crucial role in foam cell formation and LDL uptake in macrophages. However, the results of in vivo studies using gene-knockout models are somewhat different. The effect of SR-A1 on atherogenesis is controversial, whereas the proatherogenic role of CD36 has been clearly demonstrated in vivo. In 2005, Moore et al. [8] reported that defects in macrophage lipid uptake were found in apolipoprotein E (apoE) deficient mice in which either the CD36 or SR-A gene was deleted, leading to an increase in the size of atherosclerotic lesions. However, the other expanded studies showed that less atherosclerotic lesion formation in CD36 deficiency in apoE or LDLR null atherogenic mice model. Moreover, CD36 deficiency is sufficient to decrease of atherosclerosis in CD36/SR-A/apoE triple-null mice without the additional effect of SR-A1 deficiency [910]. Furthermore, a bone marrow transplantation assay also provided support for the proatherogenic role of macrophage CD36 [11]. These studies have demonstrated that macrophage CD36 promotes atherosclerosis via the uptake of modified LDL.
Cholesterol esterification
Within the macrophage, modified LDL is hydrolyzed to free cholesterol and fatty acid. Excess free cholesterol undergoes reesterification by the endoplasmic reticulum (ER)-resident protein acetyl-coenzyme A cholesterol acyltransferase 1 (ACAT1) or by sterol O-acyltransferase 1 (SOAT1), and is stored as CE in cytoplasmic lipid droplets (LDs). Although decreasing the expression or activity of ACAT1 was expected to have therapeutic effects through inhibition of foam cell formation, both ACAT1/apoE and ACAT1/LDLR double-null mice have been found to show similarly sized or only slightly smaller atherosclerotic lesions than controls [1213]. Furthermore, the efficacy of ACAT inhibitors in clinically preventing atherosclerosis in humans has not been successfully demonstrated [14]. Thus, these studies have clarified that cholesterol esterification is a passive protective response to excess free cholesterol when cholesterol efflux pathways are saturated.
MACROPHAGE CHOLESTEROL EFFLUX
Neutral cholesterol lipolysis
The hydrolysis of intracellular CE is the initial step of cholesterol efflux in macrophages. Because CE hydrolysis, which precedes cholesterol efflux, occurs in the cytoplasm at neutral pH levels, the catalyze enzymes have been collectively called neutral CE hydrolase. Three enzymes have been proposed to be components of neutral CE hydrolase in macrophages: hormone-sensitive lipase (HSL) [15]; carboxylesterase 1 (CEH or CES1), the human homolog of the murine triacylglycerol hydrolase [16]; and neutral cholesterol ester hydrolase 1 (NCEH1), which is also known as KIAA1363 or arylacetamide deacetylase like 1 (AADACL1) [17]. However, their importance has yet to be fully elucidated. Nevertheless, a common feature of all of the studies that have been conducted on this topic is that enhancing LD-related CE hydrolysis reduces CE accumulation and improves cholesterol efflux; thereby, reducing arteriosclerosis [1819]. Thus, these studies demonstrate that CE hydrolysis in macrophage cholesterol efflux is rate-limiting, and show that it is important to clarify the mechanisms mediating CE hydrolysis in foam cells for the treatment of atherogenesis.
Acidic cholesterol lipolysis and lipophagy
In 1999, Avart et al. [20] firstly discovered in vitro that a lysosomal component is involved in CE hydrolysis in foam cells. In this process, CE is hydrolyzed by lysosomal acid lipase (LAL), a lysosomal cholesterol esterase that exhibits optimal activity at the acidic pH of the lysosomal lumen. Recently, Ouimet et al. [21] reported that under lipid-loading conditions, autophagy mediated the delivery of cytoplasmic LDs to lysosomes in macrophages and that LAL in the lysosomal lumen hydrolyzed LD CE to generate free cholesterol for efflux. Ouimet et al. [21] also found autophagy to be specifically induced in response to atherogenic lipoprotein accumulation within macrophages, and thus, concluded that the autophagy-LAL pathway is a critical contributor to the mobilization of LD-associated CE for RCT.
Autophagy is a conserved cellular process for the natural breakdown of unnecessary or non-functional cellular organelles and proteins by fusion with lysosomal compartments [22]. Autophagy is highly inducible under environmental stresses such as starvation and oxidative stress, and it plays an important role in maintaining essential cellular functions and protecting against infections with pathogens [2324]. In starvation conditions, autophagy promotes the degradation of cytoplasmic components non-selectively, whereas during nutrient-rich conditions, autophagy selectively eliminates specific cytoplasmic cargo. Depending upon the loaded and digested cytoplasmic cargo, autophagy has been divided into aggrephagy, mitophagy, pexophagy, ER-phagy, xenophagy, and so on [25]. Recently, an alternative pathway of LD degradation through the lysosomal pathway of autophagy has been described and termed lipophagy [26].
Lipophagy was originally described in hepatocytes, where it is critical for maintaining cellular energy homeostasis in obesity and metabolic syndrome. In vitro and in vivo studies have demonstrated the selective uptake of LDs by autophagosomes, and the genetic or chemical inhibition of autophagy has been shown to reduce the β-oxidation of free fatty acids due to the increased accumulation of lipids and LDs [23]. Importantly, Singh and Cuervo [23] found that impaired autophagy in cultured hepatocytes and mouse liver led to abnormally high levels of hepatic cholesterol along with aberrant triacylglycerol deposition because of the defective clearance of LDs. Furthermore, that study identified a previously unknown function for autophagy in lipid metabolism, with possible implications for various human diseases involving lipid over-accumulation, such as atherosclerosis and cardiovascular disease. Indeed, another report demonstrated that lipophagy became dysfunctional in atherogenesis, and that its deficiency promoted atherosclerosis in part through inflammasome hyperactivation caused by cholesterol crystal accumulation in macrophages [27]. Additionally, macrophage-specific autophagy deficiency led to increased apoptosis and oxidative stress in advanced lesional macrophages, promoted plaque necrosis, and worsened lesional efferocytosis [28]. Thus, the autophagy pathway may contribute to regulate access to lipid stimulation in macrophages in atherosclerotic plaques.
Consistent with these findings, LAL has been the focus of new studies. LAL is the central enzyme for hydrolysis in lysosomes, and its deficiency leads to human cholesterol storage disorders such as CE storage disease (CESD) and Wolman disease. Bowden et al. [29] provided an explanation of the hypolipoproteinemia seen in CESD patients by showing that LAL activity contributed to the regulation of ATP-binding cassette subfamily A member 1 (ABCA1) expression and activity. Additional studies will be required to clarify the mechanism of LAL, but it is clear that LAL plays an essential role in atherosclerosis via acid lipolysis.
MACROPHAGE CHOLESTEROL EFFLUX
This macrophage cholesterol efflux function is predominantly mediated by high density lipoprotein (HDL). Apolipoprotein A1 (apoA1), the most abundant protein in HDL, and mature HDL particles serve as acceptors of macrophage cholesterol efflux. Lipid-poor apoA1 promotes the efflux of cholesterol from macrophages via ABCA1, and mature HDL promotes macrophage cholesterol efflux through the ATP-binding cassette subfamily G member 1 (ABCG1) transporter. Additionally mature HDL can directly deliver cholesterol to the liver via scavenger receptor class B member 1 (SR-BI), or indirectly through transfer of cholesterol to apoB-containing lipoproteins, with sequential uptake by LDL receptors in the liver [30]. Both in vitro and in vivo analyses have shown that ATP-binding cassette (ABC) transporter deficiency promotes atherosclerotic lesion development through impaired cholesterol efflux in macrophages [31323334]. Consistent with these findings, lipophagy-mediated macrophage efflux is primarily ABCA1-dependent, since cholesterol delivery to ABCA1 in the lipophagy-defective macrophages is limited by liver X receptor (LXR) [21].
LXRs are members of the nuclear receptor family of ligand-dependent transcription factors that mediate the regulation of cholesterol homeostasis [35]. LXRs form heterodimers with retinoid X receptors (RXRs) and activate transcription of specific target genes such as those coding for ABC transporters and apoE, which can function as an acceptor; thus, promoting cholesterol transport by the ABCA-1 dependent pathway [3637]. Additionally, LXRs induce the synthesis of fatty acids, which act as substrates of ACAT1 in cholesterol esterification reactions [38]. Despite these results, the relationship of autophagy with LXR and miR-33 [39], which regulates the expression of ABCA1, has yet to be clarified. Therefore, it will be necessary to elucidate the association of LXR and autophagy in macrophage cholesterol homeostasis.
CONCLUSIONS
Macrophages are essential cells that regulate lipid metabolism, especially through RCT, affecting both the progression and regression of atherosclerosis. Therefore, many studies have been conducted to establish a therapeutic strategy for atherosclerosis by studying the cholesterol pathway of macrophages. Recent studies have identified the role of lipophagy, which regulates the acidic hydrolysis of cholesterol from macrophage LDs via the lysosomes, in macrophage RCT and atherosclerosis. Understanding the effective removal of cholesterol from macrophage foam cells, in combination with future studies elucidating the role of lipophagy in macrophages, will lead to the development of novel therapeutic avenues for atherogenesis and metabolic syndrome.
ACKNOWLEDGMENTS
This work was supported by a National Research Foundation of Korea (NRF) grant funded by the Korean government (No. 2012R1A3A2026454 and 2012R1A6A3A04040206).
Notes
CONFLICTS OF INTEREST: No potential conflict of interest relevant to this article was reported.