Acquired Forms of Fibroblast Growth Factor 23-Related Hypophosphatemic Osteomalacia
Article information

Abstract
Fibroblast growth factor 23 (FGF23) is a pivotal humoral factor for the regulation of serum phosphate levels and was first identified in patients with autosomal dominant hypophosphatemic rickets and tumor-induced osteomalacia (TIO), the most common form of acquired FGF23-related hypophosphatemic rickets/osteomalacia (FGF23rHR). After the identification of FGF23, many other inherited and acquired forms of FGF23rHR were reported. In this review article, the detailed features of each acquired FGF23rHR are discussed, including TIO, ectopic FGF23 syndrome with malignancy, fibrous dysplasia/McCune-Albright syndrome, Schimmelpenning-Feuerstein-Mims syndrome/cutaneous skeletal hypophosphatemia syndrome, intravenous iron preparation-induced FGF23rHR, alcohol consumption-induced FGF23rHR, and post-kidney transplantation hypophosphatemia. Then, an approach for the differential diagnosis and therapeutic options for each disorder are concisely introduced. Currently, the majority of endocrinologists might only consider TIO when encountering patients with acquired FGF23rHR; an adequate differential diagnosis can reduce medical costs and invasive procedures such as positron emission tomography/computed tomography and venous sampling to identify FGF23-producing tumors. Furthermore, some acquired FGF23rHRs, such as intravenous iron preparation/alcohol consumption-induced FGF23rHR, require only cessation of drugs or alcohol to achieve full recovery from osteomalacia.
INTRODUCTION
Fibroblast growth factor 23 (FGF23) is a humoral factor that regulates serum inorganic phosphate (Pi) levels by modulating the expression levels of solute carrier family (SLC) 34 member 1 (SLC34A1), SLC34A3, cytochrome P450 family (CYP) 27 subfamily B member 1 (CYP27B1), and CYP24A1 in the proximal tubule via the receptor complex Klotho (KL)/FGF receptor (FGFR) 1 for FGF23 [1]. Mature osteocytes sense serum Pi levels and modulate the secretion of FGF23 via phosphorylation of the intracellular domain of FGFR1 to maintain serum phosphate levels within an adequate physiological range to prevent the development of hypophosphatemic rickets/osteomalacia and hyperphosphatemic tumoral calcinosis [2-4].
After identification of FGF23 as a pivotal serum phosphate-regulating factor in clinical cases with autosomal dominant hypophosphatemic rickets (ADHR; OMIM ID #193100) and tumor-induced osteomalacia (TIO), a variety of inherited and acquired forms of FGF23-related hypophosphatemic rickets/osteomalacia (FGF23rHR) were reported [1,5,6]. Acquired forms of FGF23rHR include TIO, ectopic FGF23 syndrome with malignancy, fibrous dysplasia (FD)/McCune-Albright syndrome (MAS) (OMIM ID #174800), Schimmelpenning-Feuerstein-Mims syndrome (SFM)/cutaneous skeletal hypophosphatemia syndrome (CSHS) (OMIM ID #163200), intravenous iron preparation-induced FGF23rHR, alcohol consumption-induced FGF23rHR, and post-kidney transplantation hypophosphatemia (Table 1). Although there are many different forms of acquired FGF23rHR, the majority of endocrinologists, rheumatologists, and orthopedists might only consider TIO when encountering patients with an acquired form of FGF23rHR, which might result in unnecessary expense and unnecessary exposure of patients to somatostatin receptor positron emission tomography/ computed tomography (PET/CT) and FGF23 venous sampling. To avoid this, clinicians should be aware of the variety of acquired FGF23rHRs and use adequate approaches to differentiate these disorders, which might also lead to simple and inexpensive treatment options without surgical or pharmaceutical intervention.

Acquired FGF23-Related Hypophosphatemic Rickets/Osteomalacia (FGF23rHR)
In this review, acquired FGF23rHR is divided into four categories: acquired FGF23rHR by neoplasms, somatic mosaicisms, exogenous factors, and other causes. Each disorder is concisely described. Furthermore, clinical steps to differentiate these disorders are presented.
ACQUIRED FGF23rHR CAUSED BY NEOPLASMS
Tumor-induced osteomalacia
In 2001, Shimada et al. [6] revealed that FGF23 is the key phosphaturic factor inappropriately secreted from a phosphaturic mesenchymal tumor (PMT) in a patient with TIO. To date, two different driver translocations, fibronectin 1 (FN1)-FGFR1 and FN1-FGF1, which are thought to downregulate the phosphate-sensing threshold in PMTs, have been identified [7,8]. In tumors without FN1-FGFR1 or FN1-FGF1 translocations, ectopic expression of KL might be involved in the mechanisms underlying the oversecretion of FGF23 due to the putative autocrine action of FGF23 stimulating the KL/FGFR1 receptor complex in identical osteocyte-like cells, though a driver variant for ectopic expression of KL has not been identified [9,10].
Intriguingly, a variety of cell lineages, including mesenchymal and hematopoietic cell lineages, are observed in PMTs and usually develop in bone or soft tissue. As expected, osteocyte-like, brand, spindle-shaped cells were identified as FGF23-producing cells, yet the mechanisms involved in sustaining the proliferation of PMTs remain unclear because FGF23-producing mature osteocytes are supposed to be terminally differentiated cells without proliferative potential [11].
Occasionally, it is difficult to identify PMTs in patients with TIO because the tumor might be small and develop in any part of the bone or soft tissue. To localize PMTs, the sensitivity and specificity of somatostatin receptor PET/CT and FGF23 venous sampling have been reported; however, even with these methods, the discovery rates of PMTs are still only 60% to 70% [12-14]. The identified tumor should be subjected to wide excision, as residual tumor tissue is associated with false biochemical remission and local recurrence [14]. For inoperable tumors, the anti-FGF23 antibody burosumab shows efficacy comparable to that of surgery, and the pan-FGFR inhibitor infigratinib has a cytoreductive effect on malignant PMTs with metastasis [15-17]. Additionally, it is recommended to inform TIO patients with PMTs preoperatively about treatment options with burosumab when the PMTs are located in regions associated with high surgical risk, permanent damage, and cosmetic problems. Clarification of the causes underlying unidentified PMTs and the development of pharmaceutical treatment for malignant PMTs with fewer adverse effects are warranted.
Ectopic FGF23 syndrome
For years, there have been many case reports on the development of hypophosphatemic osteomalacia in patients with advanced-stage lung cancer and prostate cancer and, less frequently, in patients with other types of advanced-stage malignancies, and paraneoplastic syndrome has been proposed as a mechanism [18]. In 2013, Leaf et al. [19] confirmed inappropriately elevated FGF23 in a patient with metastatic colon carcinoma and hypophosphatemic osteomalacia. In 2016, Sauder et al. [20] revealed unsuitably elevated FGF23 and ectopic FGF23 production in a metastatic lesion in the liver by immunohistochemistry in a patient with pulmonary small cell carcinoma and hypophosphatemic osteomalacia. In 2022, Hidaka et al. [14] reported that five of 24 suspected TIO patients without identified PMTs had advanced malignancies (two with lung cancer, two with prostate cancer, and one with gastric cancer). A systemic literature review revealed 53 patients with suspected ectopic FGF23 syndrome, including 26 with prostate cancer, 12 with lung cancer, four with blood cancer, three with breast cancer, two with ovarian cancer, one with gastric cancer, one with colon cancer, one with renal cancer, one with a pancreas tumor, one with a brain tumor, and one with thyroid cancer [18].
Interestingly, the majority of lung cancer patients with suspected ectopic FGF23 syndrome simultaneously present ectopic adrenocorticotropic hormone syndrome and syndrome of inappropriate antidiuretic hormone secretion [18]. To date, the driver variants for ectopic FGF23 syndrome have not been found to be the same as those in TIO (FN1-FGFR1, FN1-FGF1). Pseudofracture detected by bone scintigraphy might be misdiagnosed as a bone metastasis lesion; therefore, routine measurements of serum Pi and bone alkaline phosphatase (BAP) and adequate treatment with active vitamin D and Pi supplements or burosumab for patients with identified ectopic FGF23 are recommended to prevent pseudofractures and fractures or accelerate their healing. For patients with ectopic FGF23 syndrome with mild to moderate FGF23rHR, effective chemotherapy targeted for malignant tumors might normalize FGF23 levels.
ACQUIRED FGF23rHR CAUSED BY SOMATIC MOSAICISMS
Fibrous dysplasia/McCune-Albright syndrome
MAS is caused by gain-of-function somatic mosaicisms in the GNAS gene, which results in the development of café-au-lait spots, early puberty, FD, and other endocrine disorders accompanied by excessive hormonal activity. FD, which may develop in isolation, can cause multiple severe bone deformities and fractures due to the replacement of bone by structurally unsound fibro-osseous tissue. The extent of disease burden can be determined by bone scintigraphy, and approximately 50% of patients with FDs are thought to cause rickets/osteomalacia.
In 2003, Riminucci et al. [21] revealed that rickets/osteomalacia in patients with FD/MAS stems from the inappropriate oversecretion of FGF23 from osteocytes in the FD. Recently, low to low-normal serum Pi status was identified as a significant risk factor for the development of bone complications (in fractures, orthopedic surgeries, and scoliosis) [22]. The efficacy of burosumab was presented in a case report of a 7-year-old boy with FD/MAS [23]. To date, bone antiresorptive therapies, including bisphosphonate and denosumab, have been recommended for prevention of fractures at FD lesions. However, the use of bone antiresorptive therapy increases the risk of hypophosphatemic rickets/osteomalacia-associated low-osteoplastic fractures. Therefore, discontinuation of bone antiresorptive therapy and initiation of Pi supplementation, active vitamin D, or burosumab should be considered for FD/MAS patients with low to lownormal Pi and increased BAP.
Schimmelpenning-Feuerstein-Mims syndrome/cutaneous skeletal hypophosphatemia syndrome
SFM/CSHS (hereafter, CSHS) is characterized by multiple epidermal, melanocytic, and sebaceous nevi with hypophosphatemic rickets/osteomalacia due to somatic gain-of-function mosaicisms in RAS genes (HRAS, NRAS, and KRAS).
In 2014, Avitan-Hersh et al. [24] and Lim et al. [25] reported that rickets/osteomalacia in patients with CSHS develops through inappropriate increases in the serum FGF23 concentration. As the immunohistochemical expression of FGF23 was negative in nevi, gain-of-function RAS genes in mature osteocytes might be the source of the inappropriate secretion of FGF23 [25]. It has been revealed that serum Pi levels are monitored by mature osteocytes via phosphorylation of the intracellular domain of FGFR1 which subsequently provokes isolated phosphorylation of Y196 in FGFR substrate 2α (FRS2α). This phosphorylation of FRS2α is followed by stepwise phosphorylation of RAS, RAF, mitogen-activated extracellular signal-regulated kinase (MEK), and extracellular signal-regulated kinase (ERK) (RAS signaling) to increase early growth response 1 (EGR1) and E26 transformation-specific variant transcription factor 5 (ETV5) in the nucleus. Finally, EGR1 and ETV5 stimulate the transcription of GALNT3, and increased GALNT3 protects intact FGF23 from cleavage via O-glycosylation, which elevates serum intact FGF23 levels [2]. Therefore, it is comprehensible that some RASopathies with increased RAS signaling could develop FGF23rHR by interfering with this Pi sensing and FGF23 modulating cascade in mature osteocytes. However, the involvement of other RASopathies, including Noonan syndrome, cardio-facio-cutaneous syndrome, and Costello syndrome, has not been identified. The efficacy of burosumab was confirmed in a 15-year-old boy with CSHS; this was followed by two case reports involving an 11-year-old girl and a 20-year-old male with CSHS [26,27].
ACQUIRED FGF23rHR CAUSED BY EXOGENOUS FACTORS
Intravenous iron preparation-induced FGF23rHR
In 2009, Shimizu et al. [28] and Schouten et al. [29] reported that the intravenous administration of saccharated ferric oxide and ferric polymaltose induces FGF23rHR, respectively. Additionally, Wolf et al. [30,31] prospectively confirmed that the intravenous administration of ferric carboxymaltose causes an increase in intact FGF23 in a large cohort of patients with irondeficiency anemia, whereas iron dextran and ferumoxytol did not. Therefore, additive moieties of saccharated ferric oxide, ferric polymaltose, and ferric carboxymaltose might modulate the Pi-FGFR1-FGF23 axis through unidentified mechanisms that facilitate O-glycosylation of the FGF23 protein independent of the serum Pi concentration in FGF23rHR development. Intravenous iron preparation-induced FGF23rHR is not commonly observed because intravenous iron preparation is generally used in patients receiving hemodialysis, in whom FGF23 has no effect or has little effect on the proximal tubule and is already extremely elevated [32]. Iron-deficient patients without chronic kidney disease (CKD) G5/5D might develop intravenous iron preparation-induced FGF23rHR when they are treated with the abovementioned drugs due to intolerance of the oral iron preparation or difficulty in oral administration because of severe illness or certain digestive tract disorders, such as advanced esophageal cancer or Crohn’s disease. In these patients, it is recommended to use iron dextran and ferumoxytol, which have no hypophosphatemic effect, or ferric derisomaltose, which has a mild hypophosphatemic effect [33,34].
Alcohol consumption-induced FGF23rHR
In 2021, Hidaka et al. [35] studied 43-year-old and 60-year-old male patients who developed acquired FGF23rHR; their serum FGF23 levels and Pi levels were, very intriguingly, normalized during cessation of alcohol consumption. Given the substantial proportion of the population that consumes alcohol, alcohol consumption-induced FGF23rHR is extremely rare, and the underlying mechanism has not been proven. However, cessation of alcohol consumption for 1 to 2 months is recommended to exclude this condition before implementing somatostatin receptor PET/CT or FGF23 venous sampling when presuming TIO.
ACQUIRED FGF23rHR WITH OTHER CAUSES
Post-kidney transplantation hypophosphatemia
After kidney transplantation, the majority of patients experience transient hypophosphatemia for approximately 3 to 18 months, which can be attributed to secondary or tertiary hyperparathyroidism. However, these patients include those with normal parathyroid hormone levels or those whose hyperparathyroidism has normalized before recovery of hypophosphatemia.
In 2006, Bhan et al. [36] reported that the mechanism underlying post-kidney transplantation hypophosphatemia is consistent with high serum FGF23 levels. Nevertheless, the development of osteomalacia after kidney transplantation is extremely rare, possibly because a longer period with hypophosphatemia is needed to develop into osteomalacia. Therefore, treatment for transient hypophosphatemia might be avoided. In particular, Pi supplementation should not be applied, as it is associated with the progression of CKD.
DIFFERENTIAL DIAGNOSIS OF ACQUIRED FGF23rHR
A flowchart for the differential diagnosis of acquired FGF23rHR is suggested in Fig. 1. It is of clinical importance that causes for acquired FGF23rHR other than TIO should be strictly excluded before conducting somatostatin receptor PET/CT, FGF23 venous sampling, and surgery to remove suspected PMTs to save medical costs and avoid unnecessary investigational procedures, surgery, and pharmaceutical therapy.
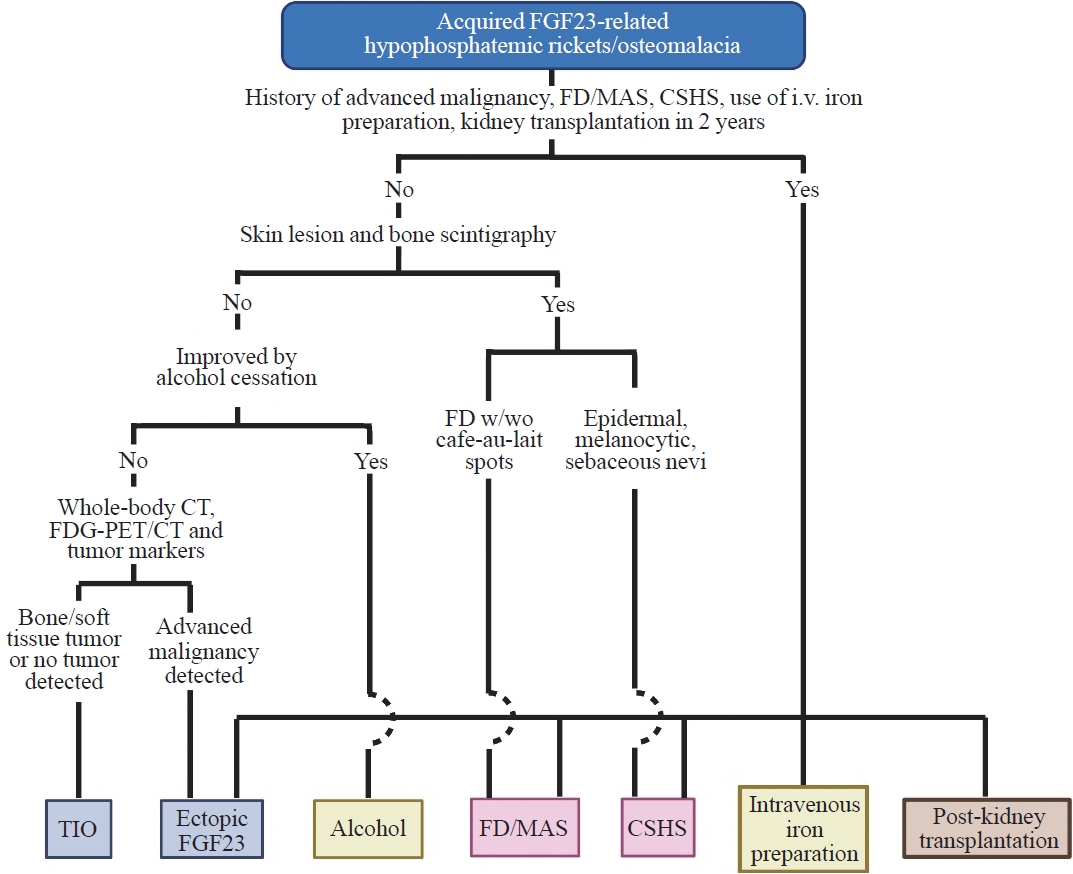
Differential diagnosis of acquired fibroblast growth factor 23 (FGF23)-related hypophosphatemic rickets/osteomalacia. FD, fibrous dysplasia; MAS, McCune-Albright syndrome; CSHS, cutaneous skeletal hypophosphatemia syndrome; i.v., intravenous drip; w/wo, with or without; FDG, fluorodeoxyglucose; PET/CT, positron emission tomography/computed tomography; TIO, tumor-induced osteomalacia.
The flowchart starts with a medical history of advanced malignancies; FD/MAS and CSHS; intravenous iron preparation; and kidney transplantation within 2 years. Next, skin lesions, including café-au-lait spots, epiderma, melanocytic, and sebaceous nevi, and bone FD should be confirmed by inspection and bone scintigraphy to exclude FD/MAS and CSHS. Subsequently, to exclude alcohol consumption-induced FGF23rHR, cessation of alcohol should be considered in appropriate patients. Finally, whole-body CT, FDG-PET/CT, and tumor marker data assessment are recommended to detect advanced malignancies, especially lung and prostate cancers. The exploration of PMT by methods such as somatostatin receptor PET/CT and FGF23 venous sampling becomes acceptable after all these diagnostic exclusion steps.
CONCLUSIONS
Since the identification of FGF23 as a pathological phosphaturic factor in a patient with TIO in 2001, several other acquired patterns of FGF23rHR have been identified, as FGF23 plays a pivotal role in the regulation of serum Pi. Clinicians involved in the diagnosis and treatment of FGF23rHR are advised to note that other causes of acquired FGF23rHR should be strictly excluded before diagnosing TIO to avoid unnecessary and invasive investigations, surgery, and pharmaceutical therapy.
Notes
CONFLICTS OF INTEREST
Nobuaki Ito receives research support from Kyowa Kirin Co. Ltd. All other authors have nothing to disclose.