
Search
- Page Path
- HOME > Search
Original Article
- Diabetes, obesity and metabolism
- Docosahexanoic Acid Attenuates Palmitate-Induced Apoptosis by Autophagy Upregulation via GPR120/mTOR Axis in Insulin-Secreting Cells
- Seok-Woo Hong, Jinmi Lee, Sun Joon Moon, Hyemi Kwon, Se Eun Park, Eun-Jung Rhee, Won-Young Lee
- Endocrinol Metab. 2024;39(2):353-363. Published online January 23, 2024
- DOI: https://doi.org/10.3803/EnM.2023.1809

- 903 View
- 40 Download
-
 Abstract
Abstract
 PDF
PDF Supplementary Material
Supplementary Material PubReader
PubReader  ePub
ePub - Background
Polyunsaturated fatty acids (PUFAs) reportedly have protective effects on pancreatic β-cells; however, the underlying mechanisms are unknown.
Methods
To investigate the cellular mechanism of PUFA-induced cell protection, mouse insulinoma 6 (MIN6) cells were cultured with palmitic acid (PA) and/or docosahexaenoic acid (DHA), and alterations in cellular signaling and apoptosis were examined.
Results
DHA treatment remarkably repressed caspase-3 cleavage and terminal deoxynucleotidyl transferase-mediated UTP nick end labeling (TUNEL)-positive red dot signals in PA-treated MIN6 cells, with upregulation of autophagy, an increase in microtubule- associated protein 1-light chain 3 (LC3)-II, autophagy-related 5 (Atg5), and decreased p62. Upstream factors involved in autophagy regulation (Beclin-1, unc51 like autophagy activating kinase 1 [ULK1], phosphorylated mammalian target of rapamycin [mTOR], and protein kinase B) were also altered by DHA treatment. DHA specifically induced phosphorylation on S2448 in mTOR; however, phosphorylation on S2481 decreased. The role of G protein-coupled receptor 120 (GPR120) in the effect of DHA was demonstrated using a GPR120 agonist and antagonist. Additional treatment with AH7614, a GPR120 antagonist, significantly attenuated DHA-induced autophagy and protection. Taken together, DHA-induced autophagy activation with protection against PA-induced apoptosis mediated by the GPR120/mTOR axis.
Conclusion
These findings indicate that DHA has therapeutic effects on PA-induced pancreatic β-cells, and that the cellular mechanism of β-cell protection by DHA may be a new research target with potential pharmacotherapeutic implications in β-cell protection.

Namgok Lecture 2022
- Diabetes, Obesity and Metabolism
- Incretin and Pancreatic β-Cell Function in Patients with Type 2 Diabetes
- Chang Ho Ahn, Tae Jung Oh, Se Hee Min, Young Min Cho
- Endocrinol Metab. 2023;38(1):1-9. Published online February 13, 2023
- DOI: https://doi.org/10.3803/EnM.2023.103
- 3,335 View
- 362 Download
- 1 Web of Science
- 1 Crossref
-
Abstract
PDFPubReader ePub
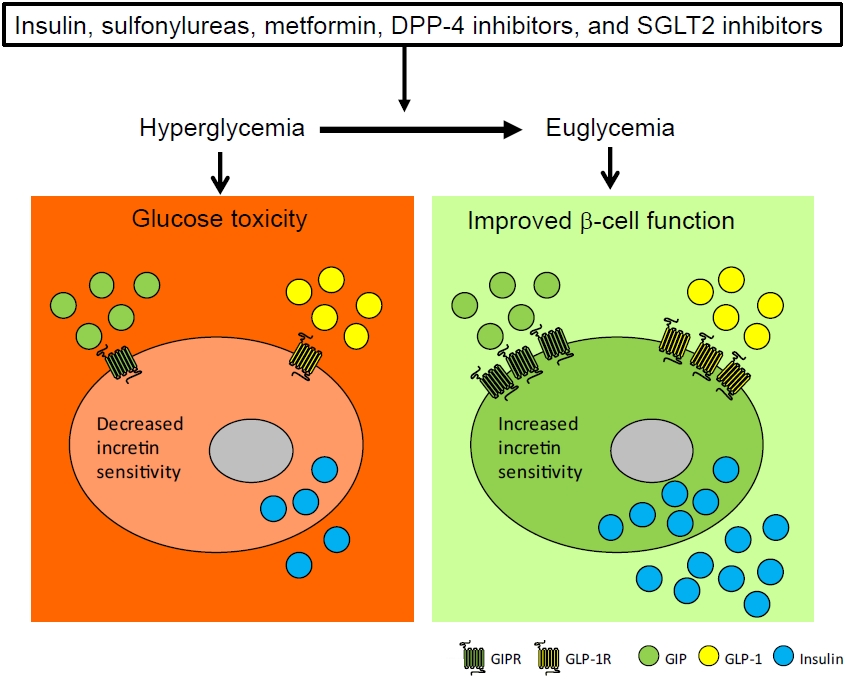
- To maintain normal glucose homeostasis after a meal, it is essential to secrete an adequate amount of insulin from pancreatic β-cells. However, if pancreatic β-cells solely depended on the blood glucose level for insulin secretion, a surge in blood glucose levels would be inevitable after the ingestion of a large amount of carbohydrates. To avoid a deluge of glucose in the bloodstream after a large carbohydrate- rich meal, enteroendocrine cells detect the amount of nutrient absorption from the gut lumen and secrete incretin hormones at scale. Since insulin secretion in response to incretin hormones occurs only in a hyperglycemic milieu, pancreatic β-cells can secrete a “Goldilocks” amount of insulin (i.e., not too much and not too little) to keep the blood glucose level in the normal range. In this regard, pancreatic β-cell sensitivity to glucose and incretin hormones is crucial for maintaining normal glucose homeostasis. In this Namgok lecture 2022, we review the effects of current anti-diabetic medications on pancreatic β-cell sensitivity to glucose and incretin hormones.
-
Citations
Citations to this article as recorded by
- Initial Combination Therapy in Type 2 Diabetes
Ji Yoon Kim, Nam Hoon Kim
Endocrinology and Metabolism.2024; 39(1): 23. CrossRef
- Initial Combination Therapy in Type 2 Diabetes
Original Articles
- Endocrine Research
- Clusterin Protects Lipotoxicity-Induced Apoptosis via Upregulation of Autophagy in Insulin-Secreting Cells
- Seok-Woo Hong, Jinmi Lee, Min Jeong Kim, Sun Joon Moon, Hyemi Kwon, Se Eun Park, Eun-Jung Rhee, Won-Young Lee
- Endocrinol Metab. 2020;35(4):943-953. Published online December 2, 2020
- DOI: https://doi.org/10.3803/EnM.2020.768
- 5,663 View
- 135 Download
- 4 Web of Science
- 6 Crossref
-
Abstract
PDFSupplementary MaterialPubReader ePub
- Background
There is a great need to discover factors that could protect pancreatic β-cells from apoptosis and thus prevent diabetes mellitus. Clusterin (CLU), a chaperone protein, plays an important role in cell protection in numerous cells and is involved in various cellular mechanisms, including autophagy. In the present study, we investigated the protective role of CLU through autophagy regulation in pancreatic β-cells.
Methods
To identify the protective role of CLU, mouse insulinoma 6 (MIN6) cells were incubated with CLU and/or free fatty acid (FFA) palmitate, and cellular apoptosis and autophagy were examined.
Results
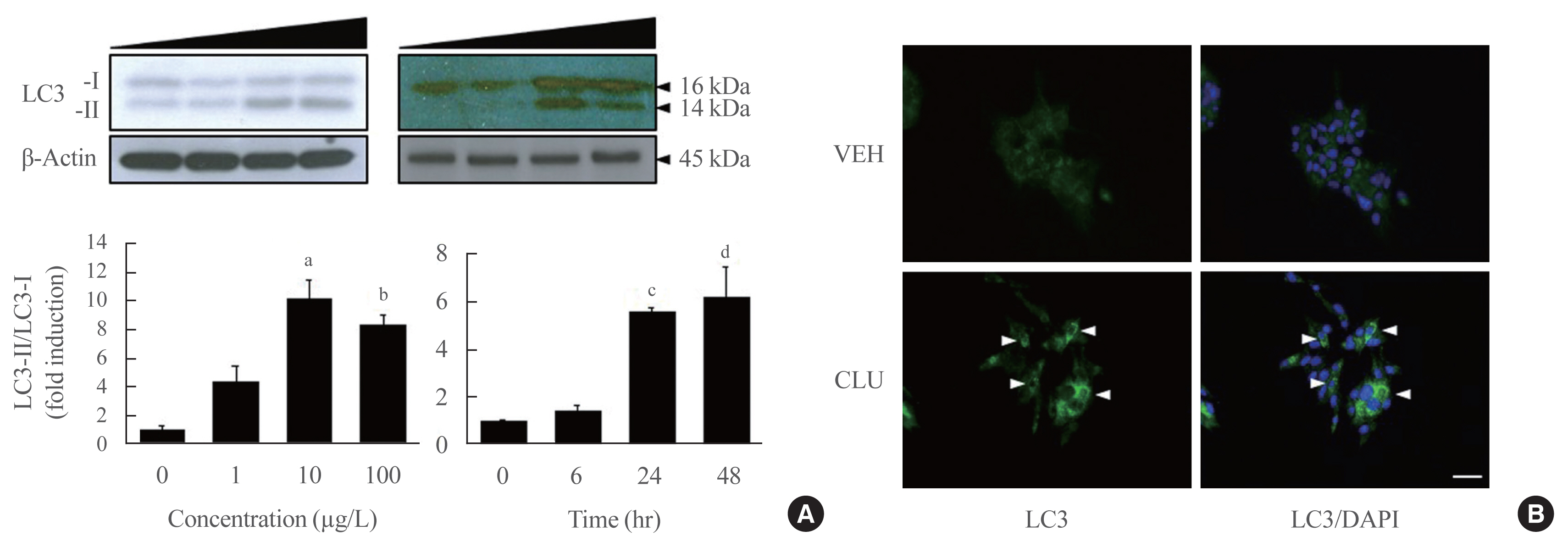
Treatment with CLU remarkably upregulated microtubule-associated protein 1-light chain 3 (LC3)-II conversion in a doseand time-dependent manner with a significant increase in the autophagy-related 3 (Atg3) gene expression level, which is a mediator of LC3-II conversion. Moreover, co-immunoprecipitation and fluorescence microscopy experiments showed that the molecular interaction of LC3 with Atg3 and p62 was markedly increased by CLU. Stimulation of LC3-II conversion by CLU persisted in lipotoxic conditions, and FFA-induced apoptosis and dysfunction were simultaneously improved by CLU treatment. Finally, inhibition of LC3-II conversion by Atg3 gene knockdown markedly attenuated the cytoprotective effect of CLU.
Conclusion
Taken together, these findings suggest that CLU protects pancreatic β-cells against lipotoxicity-induced apoptosis via autophagy stimulation mediated by facilitating LC3-II conversion. Thus, CLU has therapeutic effects on FFA-induced pancreatic β-cell dysfunction. -
Citations
Citations to this article as recorded by- Exercise as a non-pharmacological intervention to protect pancreatic beta cells in individuals with type 1 and type 2 diabetes
Alexandra Coomans de Brachène, Corentin Scoubeau, Anyïshai E. Musuaya, Jose Maria Costa-Junior, Angela Castela, Julie Carpentier, Vitalie Faoro, Malgorzata Klass, Miriam Cnop, Decio L. Eizirik
Diabetologia.2023; 66(3): 450. CrossRef - Apolipoprotein J Attenuates Vascular Restenosis by Promoting Autophagy and Inhibiting the Proliferation and Migration of Vascular Smooth Muscle Cells
Ning Yang, Bo Dong, Yanqiu Song, Yang Li, Lu Kou, Qin Qin
Journal of Cardiovascular Translational Research.2022; 15(5): 1086. CrossRef - Targets for rescue from fatty acid-induced lipotoxicity in pancreatic beta cells
Seok-Woo Hong, Won-Young Lee
Cardiovascular Prevention and Pharmacotherapy.2022; 4(2): 57. CrossRef - Co-regulators of autophagy and the cell cycle in HFD − As treated mice
Marzieh Zeinvand-Lorestani, Mohammad Javad Khodayar, Ali Teimoori, Najmaldin Saki, Akram Ahangarpour, Ali Ranjbar, Hamed Zeinvand-Lorestani
Journal of Trace Elements and Minerals.2022; 2: 100018. CrossRef - Targeting pancreatic β cells for diabetes treatment
Chirag Jain, Ansarullah, Sara Bilekova, Heiko Lickert
Nature Metabolism.2022; 4(9): 1097. CrossRef - Mechanisms of Beta-Cell Apoptosis in Type 2 Diabetes-Prone Situations and Potential Protection by GLP-1-Based Therapies
Safia Costes, Gyslaine Bertrand, Magalie A. Ravier
International Journal of Molecular Sciences.2021; 22(10): 5303. CrossRef
- Exercise as a non-pharmacological intervention to protect pancreatic beta cells in individuals with type 1 and type 2 diabetes
- Endocrine Research
- Deficiency of Sphingosine-1-Phosphate Reduces the Expression of Prohibitin and Causes β-Cell Impairment via Mitochondrial Dysregulation
- Seok-Woo Hong, Jinmi Lee, Hyemi Kwon, Se Eun Park, Eun-Jung Rhee, Cheol-Young Park, Ki-Won Oh, Sung-Woo Park, Won-Young Lee
- Endocrinol Metab. 2018;33(3):403-412. Published online September 18, 2018
- DOI: https://doi.org/10.3803/EnM.2018.33.3.403
- 4,205 View
- 50 Download
- 16 Web of Science
- 16 Crossref
-
Abstract
PDFPubReader ePub
Background Emerging evidence suggests that sphingolipids may be involved in type 2 diabetes. However, the exact signaling defect through which disordered sphingolipid metabolism induces β-cell dysfunction remains unknown. The current study demonstrated that sphingosine-1-phosphate (S1P), the product of sphingosine kinase (SphK), is an essential factor for maintaining β-cell function and survival via regulation of mitochondrial action, as mediated by prohibitin (PHB).
Methods We examined β-cell function and viability, as measured by mitochondrial function, in mouse insulinoma 6 (MIN6) cells in response to manipulation of cellular S1P and PHB levels.
Results Lack of S1P induced by sphingosine kinase inhibitor (SphKi) treatment caused β-cell dysfunction and apoptosis, with repression of mitochondrial function shown by decreases in cellular adenosine triphosphate content, the oxygen consumption rate, the expression of oxidative phosphorylation complexes, the mitochondrial membrane potential, and the expression of key regulators of mitochondrial dynamics (mitochondrial dynamin-like GTPase [OPA1] and mitofusin 1 [MFN1]). Supplementation of S1P led to the recovery of mitochondrial function and greatly improved β-cell function and viability. Knockdown of SphK2 using small interfering RNA induced mitochondrial dysfunction, decreased glucose-stimulated insulin secretion (GSIS), and reduced the expression of PHB, an essential regulator of mitochondrial metabolism. PHB deficiency significantly reduced GSIS and induced mitochondrial dysfunction, and co-treatment with S1P did not reverse these trends.
Conclusion Altogether, these data suggest that S1P is an essential factor in the maintenance of β-cell function and survival through its regulation of mitochondrial action and PHB expression.
-
Citations
Citations to this article as recorded by- Mitochondrial Cristae Morphology Reflecting Metabolism, Superoxide Formation, Redox Homeostasis, and Pathology
Petr Ježek, Martin Jabůrek, Blanka Holendová, Hana Engstová, Andrea Dlasková
Antioxidants & Redox Signaling.2023; 39(10-12): 635. CrossRef - Sphingolipids in mitochondria—from function to disease
Maryam Jamil, Lauren Ashley Cowart
Frontiers in Cell and Developmental Biology.2023;[Epub] CrossRef - Sphingosine‐1‐phosphate in mitochondrial function and metabolic diseases
Meng Duan, Pan Gao, Sheng‐xi Chen, Petr Novák, Kai Yin, Xiao Zhu
Obesity Reviews.2022;[Epub] CrossRef - Involvement of miR‐27a‐3p in diabetic nephropathy via affecting renal fibrosis, mitochondrial dysfunction, and endoplasmic reticulum stress
Lina Wu, Qingzhu Wang, Feng Guo, Xiaojun Ma, Jiao Wang, Yanyan Zhao, Yushan Yan, Guijun Qin
Journal of Cellular Physiology.2021; 236(2): 1454. CrossRef - Sphingosine‐1‐phosphate in acute exercise and training
Katarzyna Hodun, Adrian Chabowski, Marcin Baranowski
Scandinavian Journal of Medicine & Science in Sports.2021; 31(5): 945. CrossRef - The Ethyl Acetate Extract From Celastrus orbiculatus Promotes Apoptosis of Gastric Cancer Cells Through Mitochondria Regulation by PHB
Lide Tao, Zixin Yin, Tengyang Ni, Zewen Chu, Shihua Hao, Zeyu Wang, Masataka Sunagawa, Haibo Wang, Yanqing Liu
Frontiers in Pharmacology.2021;[Epub] CrossRef - Sphingosine 1-phosphate Stimulates Insulin Secretion and Improves Cell Survival by Blocking Voltage-dependent K+ Channels in β Cells
Zhihong Liu, Huanhuan Yang, Linping Zhi, Huan Xue, Zhihong Lu, Yanli Zhao, Lijuan Cui, Tao Liu, Shouan Ren, Peifeng He, Yunfeng Liu, Yi Zhang
Frontiers in Pharmacology.2021;[Epub] CrossRef - Sphingosine-1 Phosphate Lyase Regulates Sensitivity of Pancreatic Beta-Cells to Lipotoxicity
Yadi Tang, Thomas Plötz, Markus H. Gräler, Ewa Gurgul-Convey
International Journal of Molecular Sciences.2021; 22(19): 10893. CrossRef - Sphingolipids and Mitochondrial Dynamic
Lais Brigliadori Fugio, Fernanda B. Coeli-Lacchini, Andréia Machado Leopoldino
Cells.2020; 9(3): 581. CrossRef - Diminished Sphingolipid Metabolism, a Hallmark of Future Type 2 Diabetes Pathogenesis, Is Linked to Pancreatic β Cell Dysfunction
Saifur R. Khan, Yousef Manialawy, Andreea Obersterescu, Brian J. Cox, Erica P. Gunderson, Michael B. Wheeler
iScience.2020; 23(10): 101566. CrossRef - Neuronal Metabolism and Neuroprotection: Neuroprotective Effect of Fingolimod on Menadione-Induced Mitochondrial Damage
Antonio Gil, Elisa Martín-Montañez, Nadia Valverde, Estrella Lara, Federica Boraldi, Silvia Claros, Silvana-Yanina Romero-Zerbo, Oscar Fernández, Jose Pavia, Maria Garcia-Fernandez
Cells.2020; 10(1): 34. CrossRef - WITHDRAWN: Ceramide and Sphingosine 1-Phosphate in adipose dysfunction
Zijian Fang, Susan Pyne, Nigel J. Pyne
Progress in Lipid Research.2019; : 100991. CrossRef - Dynamic of mitochondrial network, cristae, and mitochondrial nucleoids in pancreatic β-cells
Petr Ježek, Andrea Dlasková
Mitochondrion.2019; 49: 245. CrossRef - Sphingosine kinase 1 overexpression induces MFN2 fragmentation and alters mitochondrial matrix Ca2+ handling in HeLa cells
I. Pulli, C. Löf, T. Blom, M.Y. Asghar, T. Lassila, N. Bäck, K.-L. Lin, J.H. Nyström, K. Kemppainen, D.M. Toivola, E. Dufour, A. Sanz, H.M. Cooper, J.B. Parys, K. Törnquist
Biochimica et Biophysica Acta (BBA) - Molecular Cell Research.2019; 1866(9): 1475. CrossRef - Ceramide and sphingosine 1-phosphate in adipose dysfunction
Zijian Fang, Susan Pyne, Nigel J. Pyne
Progress in Lipid Research.2019; 74: 145. CrossRef - S1P/S1P Receptor Signaling in Neuromuscolar Disorders
Elisabetta Meacci, Mercedes Garcia-Gil
International Journal of Molecular Sciences.2019; 20(24): 6364. CrossRef
- Mitochondrial Cristae Morphology Reflecting Metabolism, Superoxide Formation, Redox Homeostasis, and Pathology
Review Article
- Diabetes
- Pathophysiology of Type 2 Diabetes in Koreans
- Soo Heon Kwak, Kyong Soo Park
- Endocrinol Metab. 2018;33(1):9-16. Published online March 21, 2018
- DOI: https://doi.org/10.3803/EnM.2018.33.1.9
- 5,418 View
- 111 Download
- 12 Web of Science
- 10 Crossref
-
Abstract
PDFPubReader ePub
The pathophysiology of type 2 diabetes is characterized by variable degrees of insulin resistance and impaired insulin secretion. Both genetic and environmental factors serve as etiologic factors. Recent genetic studies have identified at least 83 variants associated with diabetes. A significant number of these loci are thought to be involved in insulin secretion, either through β-cell development or β-cell dysfunction. Environmental factors have changed rapidly during the past half century, and the increased prevalence of obesity and diabetes can be attributed to these changes. Environmental factors may affect epigenetic changes and alter susceptibility to diabetes. A recent epidemiologic study revealed that Korean patients with type 2 diabetes already had impaired insulin secretion and insulin resistance 10 years before the onset of diabetes. Those who developed diabetes showed impaired β-cell compensation with an abrupt decrease in insulin secretion during the last 2 years before diabetes developed. The retrograde trajectory of the disposition index differed according to the baseline subgroups of insulin secretion and insulin sensitivity. We hope that obtaining a more detailed understanding of the perturbations in the major pathophysiologic process of diabetes on the individual level will eventually lead to the implementation of precision medicine and improved patient outcomes.
-
Citations
Citations to this article as recorded by- Stress-Reducing Psychological Interventions as Adjuvant Therapies for
Diabetic Chronic Wounds
Isadora Pombeiro, João Moura, M. Graça Pereira, Eugénia Carvalho
Current Diabetes Reviews.2022;[Epub] CrossRef - Umbilical Cord-Mesenchymal Stem Cell-Conditioned Medium Improves Insulin Resistance in C2C12 Cell
Kyung-Soo Kim, Yeon Kyung Choi, Mi Jin Kim, Jung Wook Hwang, Kyunghoon Min, Sang Youn Jung, Soo-Kyung Kim, Yong-Soo Choi, Yong-Wook Cho
Diabetes & Metabolism Journal.2021; 45(2): 260. CrossRef - Dose-Dependent Effect of Smoking on Risk of Diabetes Remains after Smoking Cessation: A Nationwide Population-Based Cohort Study in Korea
Se Eun Park, Mi Hae Seo, Jung-Hwan Cho, Hyemi Kwon, Yang-Hyun Kim, Kyung-Do Han, Jin-Hyung Jung, Yong-Gyu Park, Eun-Jung Rhee, Won-Young Lee
Diabetes & Metabolism Journal.2021; 45(4): 539. CrossRef - DNA Methylation Changes Associated With Type 2 Diabetes and Diabetic Kidney Disease in an East Asian Population
Hakyung Kim, Jae Hyun Bae, Kyong Soo Park, Joohon Sung, Soo Heon Kwak
The Journal of Clinical Endocrinology & Metabolism.2021; 106(10): e3837. CrossRef - Associations among Obesity Degree, Glycemic Status, and Risk of Heart Failure in 9,720,220 Korean Adults
Eun-Jung Rhee, Hyemi Kwon, Se Eun Park, Kyung-Do Han, Yong-Gyu Park, Yang-Hyun Kim, Won-Young Lee
Diabetes & Metabolism Journal.2020; 44(4): 592. CrossRef - Smoking as a Target for Prevention of Diabetes
Ye Seul Yang, Tae Seo Sohn
Diabetes & Metabolism Journal.2020; 44(3): 402. CrossRef - Clinical characteristics of diabetic ketoacidosis in users and non-users of SGLT2 inhibitors
J.Y. Jeon, S.-K. Kim, K.-S. Kim, S.O. Song, J.-S. Yun, B.-Y. Kim, C.-H. Kim, S.O. Park, S. Hong, D.H. Seo, J.A. Seo, J.H. Noh, D.J. Kim
Diabetes & Metabolism.2019; 45(5): 453. CrossRef - Identifying Pathogenic Variants of Monogenic Diabetes Using Targeted Panel Sequencing in an East Asian Population
Seung Shin Park, Se Song Jang, Chang Ho Ahn, Jung Hee Kim, Hye Seung Jung, Young Min Cho, Young Ah Lee, Choong Ho Shin, Jong Hee Chae, Jae Hyun Kim, Sung Hee Choi, Hak C Jang, Jee Cheol Bae, Jong Cheol Won, Sung-Hoon Kim, Jong-Il Kim, Soo Heon Kwak, Kyong
The Journal of Clinical Endocrinology & Metabolism.2019; 104(9): 4188. CrossRef - Epigenetic Markers and Microbiota/Metabolite-Induced Epigenetic Modifications in the Pathogenesis of Obesity, Metabolic Syndrome, Type 2 Diabetes, and Non-alcoholic Fatty Liver Disease
Daniela Stols-Gonçalves, Luca Schiliró Tristão, Peter Henneman, Max Nieuwdorp
Current Diabetes Reports.2019;[Epub] CrossRef - The cut-off values of surrogate measures for insulin resistance in the Korean population according to the Korean Genome and Epidemiology Study (KOGES)
Bongyoung Kim, Hyun Young Choi, Wonhee Kim, Chiwon Ahn, Juncheol Lee, Jae Guk Kim, Jihoon Kim, Hyungoo Shin, Jae Myung Yu, Shinje Moon, Taulant Muka
PLOS ONE.2018; 13(11): e0206994. CrossRef
- Stress-Reducing Psychological Interventions as Adjuvant Therapies for
Diabetic Chronic Wounds
Original Articles
- Diabetes
- Pioglitazone Attenuates Palmitate-Induced Inflammation and Endoplasmic Reticulum Stress in Pancreatic β-Cells
- Seok-Woo Hong, Jinmi Lee, Jung Hwan Cho, Hyemi Kwon, Se Eun Park, Eun-Jung Rhee, Cheol-Young Park, Ki-Won Oh, Sung-Woo Park, Won-Young Lee
- Endocrinol Metab. 2018;33(1):105-113. Published online March 21, 2018
- DOI: https://doi.org/10.3803/EnM.2018.33.1.105
- 6,249 View
- 96 Download
- 19 Web of Science
- 23 Crossref
-
Abstract
PDFPubReader ePub
Background The nuclear receptor peroxisome proliferator-activator gamma (PPARγ) is a useful therapeutic target for obesity and diabetes, but its role in protecting β-cell function and viability is unclear.
Methods To identify the potential functions of PPARγ in β-cells, we treated mouse insulinoma 6 (MIN6) cells with the PPARγ agonist pioglitazone in conditions of lipotoxicity, endoplasmic reticulum (ER) stress, and inflammation.
Results Palmitate-treated cells incubated with pioglitazone exhibited significant improvements in glucose-stimulated insulin secretion and the repression of apoptosis, as shown by decreased caspase-3 cleavage and poly (adenosine diphosphate [ADP]-ribose) polymerase activity. Pioglitazone also reversed the palmitate-induced expression of inflammatory cytokines (tumor necrosis factor α, interleukin 6 [IL-6], and IL-1β) and ER stress markers (phosphor-eukaryotic translation initiation factor 2α, glucose-regulated protein 78 [GRP78], cleaved-activating transcription factor 6 [ATF6], and C/EBP homologous protein [CHOP]), and pioglitazone significantly attenuated inflammation and ER stress in lipopolysaccharide- or tunicamycin-treated MIN6 cells. The protective effect of pioglitazone was also tested in pancreatic islets from high-fat-fed KK-Ay mice administered 0.02% (wt/wt) pioglitazone or vehicle for 6 weeks. Pioglitazone remarkably reduced the expression of ATF6α, GRP78, and monocyte chemoattractant protein-1, prevented α-cell infiltration into the pancreatic islets, and upregulated glucose transporter 2 (Glut2) expression in β-cells. Moreover, the preservation of β-cells by pioglitazone was accompanied by a significant reduction of blood glucose levels.
Conclusion Altogether, these results support the proposal that PPARγ agonists not only suppress insulin resistance, but also prevent β-cell impairment via protection against ER stress and inflammation. The activation of PPARγ might be a new therapeutic approach for improving β-cell survival and insulin secretion in patients with diabetes mellitus
-
Citations
Citations to this article as recorded by- Nr1h4 and Thrb ameliorate ER stress and provide protection in the MPTP mouse model of Parkinson’s
Nancy Ahuja, Shalini Gupta, Rashmi Arora, Ella Bhagyaraj, Drishti Tiwari, Sumit Kumar, Pawan Gupta
Life Science Alliance.2024; 7(7): e202302416. CrossRef - Prosthetic vascular grafts engineered to combat calcification: Progress and future directions
Taylor K. Brown, Sara Alharbi, Karen J. Ho, Bin Jiang
Biotechnology and Bioengineering.2023; 120(4): 953. CrossRef - Obesity, diabetes mellitus, and cardiometabolic risk: An Obesity Medicine Association (OMA) Clinical Practice Statement (CPS) 2023
Harold Edward Bays, Shagun Bindlish, Tiffany Lowe Clayton
Obesity Pillars.2023; 5: 100056. CrossRef - Metformin promotes osteogenic differentiation and prevents hyperglycaemia-induced osteoporosis by suppressing PPARγ expression
Lifeng Zheng, Ximei Shen, Yun Xie, Hong Lian, Sunjie Yan, Shizhong Wang
Acta Biochimica et Biophysica Sinica.2023; 55(3): 394. CrossRef - Peroxisome proliferator-activated receptors as targets to treat metabolic diseases: Focus on the adipose tissue, liver, and pancreas
Henrique Souza-Tavares, Carolline Santos Miranda, Isabela Macedo Lopes Vasques-Monteiro, Cristian Sandoval, Daiana Araujo Santana-Oliveira, Flavia Maria Silva-Veiga, Aline Fernandes-da-Silva, Vanessa Souza-Mello
World Journal of Gastroenterology.2023; 29(26): 4136. CrossRef - Nicotinamide N-methyltransferase upregulation contributes to palmitate-elicited peroxisome proliferator-activated receptor transactivation in hepatocytes
Qing Song, Jun Wang, Alexandra Griffiths, Samuel Man Lee, Iredia D. Iyamu, Rong Huang, Jose Cordoba-Chacon, Zhenyuan Song
American Journal of Physiology-Cell Physiology.2023; 325(1): C29. CrossRef - The global perspective on peroxisome proliferator-activated receptor γ (PPARγ) in ectopic fat deposition: A review
Yanhao Qiu, Mailin Gan, Xingyu Wang, Tianci Liao, Qiuyang Chen, Yuhang Lei, Lei Chen, Jinyong Wang, Ye Zhao, Lili Niu, Yan Wang, Shunhua Zhang, Li Zhu, Linyuan Shen
International Journal of Biological Macromolecules.2023; 253: 127042. CrossRef - Chemical inducer of regucalcin attenuates lipopolysaccharide‐induced inflammatory responses in pancreatic MIN6 β‐cells and RAW264.7 macrophages
Tomiyasu Murata, Kazunori Hashimoto, Susumu Kohno, Chiaki Takahashi, Masayoshi Yamaguchi, Chihiro Ito, Itoigawa Masataka, Roji Kojima, Kiyomi Hikita, Norio Kaneda
FEBS Open Bio.2022; 12(1): 175. CrossRef - Targets for rescue from fatty acid-induced lipotoxicity in pancreatic beta cells
Seok-Woo Hong, Won-Young Lee
Cardiovascular Prevention and Pharmacotherapy.2022; 4(2): 57. CrossRef - Analysis of changes in the proteomic profile of porcine corpus luteum during different stages of the oestrous cycle: effects of PPAR gamma ligands
Zuzanna Kunicka, Karol Mierzejewski, Aleksandra Kurzyńska, Robert Stryiński, Jesús Mateos, Mónica Carrera, Monika Golubska, Iwona Bogacka, Xiaolong Wang
Reproduction, Fertility and Development.2022; 34(11): 776. CrossRef - Activation of PPARγ Protects Obese Mice from Acute Lung Injury by Inhibiting Endoplasmic Reticulum Stress and Promoting Mitochondrial Biogenesis
Yin Tang, Ke Wei, Ling Liu, Jingyue Ma, Siqi Wu, Wenjing Tang, Stéphane Mandard
PPAR Research.2022; 2022: 1. CrossRef - Effect of Pioglitazone on endoplasmic reticulum stress regarding in situ perfusion rat model
Vivien Telek, Luca Erlitz, Ibitamuno Caleb, Tibor Nagy, Mónika Vecsernyés, Bálint Balogh, György Sétáló, Péter Hardi, Gábor Jancsó, Ildikó Takács
Clinical Hemorheology and Microcirculation.2021; 79(2): 311. CrossRef - Inflammation in Metabolic Diseases and Insulin Resistance
Won-Young Lee
Cardiovascular Prevention and Pharmacotherapy.2021; 3(2): 31. CrossRef - Current Status of Endoplasmic Reticulum Stress in Type II Diabetes
Sagir Mustapha, Mustapha Mohammed, Ahmad Khusairi Azemi, Abubakar Ibrahim Jatau, Aishatu Shehu, Lukman Mustapha, Ibrahim Muazzamu Aliyu, Rabi’u Nuhu Danraka, Abdulbasit Amin, Auwal Adam Bala, Wan Amir Nizam Wan Ahmad, Aida Hanum Ghulam Rasool, Mohd Rais M
Molecules.2021; 26(14): 4362. CrossRef - JunD Regulates Pancreatic β-Cells Function by Altering Lipid Accumulation
Kexin Wang, Yixin Cui, Peng Lin, Zhina Yao, Yu Sun
Frontiers in Endocrinology.2021;[Epub] CrossRef - Pioglitazone even at low dosage improves NAFLD in type 2 diabetes: clinical and pathophysiological insights from a subgroup of the TOSCA.IT randomised trial
Giuseppe Della Pepa, Marco Russo, Marilena Vitale, Fabrizia Carli, Claudia Vetrani, Maria Masulli, Gabriele Riccardi, Olga Vaccaro, Amalia Gastaldelli, Angela A. Rivellese, Lutgarda Bozzetto
Diabetes Research and Clinical Practice.2021; 178: 108984. CrossRef - Radioprotective Effect of Pioglitazone Against Genotoxicity Induced by Ionizing Radiation in Healthy Human Lymphocytes
Roya Kazemi, Seyed J. Hosseinimehr
Cardiovascular & Hematological Agents in Medicinal Chemistry .2021; 19(1): 72. CrossRef - Recent Insights Into Mechanisms of β-Cell Lipo- and Glucolipotoxicity in Type 2 Diabetes
Maria Lytrivi, Anne-Laure Castell, Vincent Poitout, Miriam Cnop
Journal of Molecular Biology.2020; 432(5): 1514. CrossRef - Artemisinin and dihydroartemisinin promote β-cell apoptosis induced by palmitate via enhancing ER stress
Ke Chen, Hu Hua, Ziyang Zhu, Tong Wu, Zhanjun Jia, Qianqi Liu
Apoptosis.2020; 25(3-4): 192. CrossRef - Mechanisms of impaired pancreatic β‑cell function in high‑fat diet‑induced obese mice: The role of endoplasmic reticulum stress
Xiaoqing Yi, Xuan Cai, Sisi Wang, Yanfeng Xiao
Molecular Medicine Reports.2020;[Epub] CrossRef - Docosahexaenoic and Eicosapentaenoic Acids Prevent Altered-Muc2 Secretion Induced by Palmitic Acid by Alleviating Endoplasmic Reticulum Stress in LS174T Goblet Cells
Quentin Escoula, Sandrine Bellenger, Michel Narce, Jérôme Bellenger
Nutrients.2019; 11(9): 2179. CrossRef - PPAR-γ agonist, pioglitazone, reduced oxidative and endoplasmic reticulum stress associated with L-NAME-induced hypertension in rats
Eman Soliman, Shereen F. Behairy, Nabila N. El-maraghy, Shimaa M. Elshazly
Life Sciences.2019; 239: 117047. CrossRef - Changes of MODY signal pathway genes in the endoplasmic reticulum stress in INS-1-3 cells
Yanan Dong, Shirui Li, Wenhui Zhao, Yanlei Wang, Tingting Ge, Jianzhong Xiao, Yukun Li, Herve Le Stunff
PLOS ONE.2018; 13(6): e0198614. CrossRef
- Nr1h4 and Thrb ameliorate ER stress and provide protection in the MPTP mouse model of Parkinson’s
- Obesity and Metabolism
- Mitochondrial Complexes I and II Are More Susceptible to Autophagy Deficiency in Mouse β-Cells
- Min Joo Kim, Ok Kyong Choi, Kyung Sil Chae, Min Kyeong Kim, Jung Hee Kim, Masaaki Komatsu, Keiji Tanaka, Hakmo Lee, Sung Soo Chung, Soo Heon Kwak, Young Min Cho, Kyong Soo Park, Hye Seung Jung
- Endocrinol Metab. 2015;30(1):65-70. Published online March 27, 2015
- DOI: https://doi.org/10.3803/EnM.2015.30.1.65
- 3,958 View
- 40 Download
- 4 Web of Science
- 3 Crossref
-
Abstract
PDFPubReader
Background Damaged mitochondria are removed by autophagy. Therefore, impairment of autophagy induces the accumulation of damaged mitochondria and mitochondrial dysfunction in most mammalian cells. Here, we investigated mitochondrial function and the expression of mitochondrial complexes in autophagy-related 7 (
Atg7 )-deficient β-cells.Methods To evaluate the effect of autophagy deficiency on mitochondrial function in pancreatic β-cells, we isolated islets from
Atg7 F/F:RIP-Cre + mice and wild-type littermates. Oxygen consumption rate and intracellular adenosine 5'-triphosphate (ATP) content were measured. The expression of mitochondrial complex genes inAtg7 -deficient islets and in β-TC6 cells transfected with siAtg7 was measured by quantitative real-time polymerase chain reaction.Results Baseline oxygen consumption rate of
Atg7 -deficient islets was significantly lower than that of control islets (P <0.05). Intracellular ATP content ofAtg7 -deficient islets during glucose stimulation was also significantly lower than that of control islets (P <0.05). By Oxygraph-2k analysis, mitochondrial respiration inAtg7 -deficient islets was significantly decreased overall, although state 3 respiration and responses to antimycin A were unaffected. The mRNA levels of mitochondrial complexes I, II, III, and V inAtg7 -deficient islets were significantly lower than in control islets (P <0.05). Down-regulation ofAtg7 in β-TC6 cells also reduced the expression of complexes I and II, with marginal significance (P <0.1).Conclusion Impairment of autophagy in pancreatic β-cells suppressed the expression of some mitochondrial respiratory complexes, and may contribute to mitochondrial dysfunction. Among the complexes, I and II seem to be most vulnerable to autophagy deficiency.
-
Citations
Citations to this article as recorded by- Proteomic pathways to metabolic disease and type 2 diabetes in the pancreatic islet
Belinda Yau, Sheyda Naghiloo, Alexis Diaz-Vegas, Austin V. Carr, Julian Van Gerwen, Elise J. Needham, Dillon Jevon, Sing-Young Chen, Kyle L. Hoehn, Amanda E. Brandon, Laurence Macia, Gregory J. Cooney, Michael R. Shortreed, Lloyd M. Smith, Mark P. Keller,
iScience.2021; 24(10): 103099. CrossRef - Natural compound oblongifolin C inhibits autophagic flux, and induces apoptosis and mitochondrial dysfunction in human cholangiocarcinoma QBC939 cells
Aiqing Zhang, Wei He, Huimin Shi, Xiaodan Huang, Guozhong Ji
Molecular Medicine Reports.2016; 14(4): 3179. CrossRef - Autophagy deficiency in β cells blunts incretin-induced suppression of glucagon release from α cells
Min Joo Kim, Ok Kyong Choi, Kyung Sil Chae, Hakmo Lee, Sung Soo Chung, Dong-Sik Ham, Ji-Won Kim, Kun-Ho Yoon, Kyong Soo Park, Hye Seung Jung
Islets.2015; 7(5): e1129096. CrossRef
- Proteomic pathways to metabolic disease and type 2 diabetes in the pancreatic islet
 First
First Prev
Prev


