Articles
- Page Path
- HOME > Endocrinol Metab > Volume 39(1); 2024 > Article
-
Review ArticleAdrenal gland A Contemporary Approach to the Diagnosis and Management of Adrenal Insufficiency
 Keypoint
Keypoint
The hallmark clinical features of adrenal insufficiency include fatigue, appetite loss, unintentional weight loss, low blood pressure, and hyponatremia. The authors focus on using early morning serum cortisol and dehydroepiandrosterone sulfate for the diagnosis of adrenal insufficiency and also discuss stress dosing and cortisol in patients with critical illnesses. The authors note that a serum cortisol >14 μg/dL or DHEAS >65 μg/dL rules out adrenal insufficiency and emphasize that moderate doses of hydrocortisone are preferable to very supraphysiologic pulses for stress dosing. -
Suranut Charoensri1,2, Richard J. Auchus2,3,4

-
Endocrinology and Metabolism 2024;39(1):73-82.
DOI: https://doi.org/10.3803/EnM.2024.1894
Published online: January 22, 2024
1Division of Endocrinology and Metabolism, Department of Medicine, Faculty of Medicine, Khon Kaen University, Khon Kaen, Thailand
2Division of Metabolism, Endocrinology, and Diabetes, Department of Internal Medicine, University of Michigan, Ann Arbor, MI, USA
3Department of Pharmacology, University of Michigan, Ann Arbor, MI, USA
4Endocrinology & Metabolism Section, Medicine Service, LTC Charles S. Kettles VA Medical Center, Ann Arbor, MI, USA
- Corresponding author: Richard J. Auchus. Division of Metabolism, Endocrinology, and Diabetes, Department of Internal Medicine, University of Michigan, 1150 W Medical Center Drive, MSRB II, 5570B, Ann Arbor, MI 48109, USA Tel: +1-734-764-7764, Fax: +1-734-936-6684, E-mail: rauchus@med.umich.edu
Copyright © 2024 Korean Endocrine Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- 2,526 Views
- 248 Download
ABSTRACT
- Adrenal insufficiency (AI) can be classified into three distinct categories based on its underlying causes: primary adrenal disorders, secondary deficiencies in adrenocorticotropin, or hypothalamic suppression from external factors, most commonly glucocorticoid medications used for anti-inflammatory therapy. The hallmark clinical features of AI include fatigue, appetite loss, unintentional weight loss, low blood pressure, and hyponatremia. Individuals with primary AI additionally manifest skin hyperpigmentation, hyperkalemia, and salt craving. The diagnosis of AI is frequently delayed due to the non-specific symptoms and signs early in the disease course, which poses a significant challenge to its early detection prior to an adrenal crisis. Despite the widespread availability of lifesaving glucocorticoid medications for decades, notable challenges persist, particularly in the domains of timely diagnosis while simultaneously avoiding misdiagnosis, patient education for averting adrenal crises, and the determination of optimal replacement therapies. This article reviews recent advancements in the contemporary diagnostic strategy and approaches to optimal treatment for AI.
- Adrenal insufficiency (AI) is a medical condition characterized by adrenal cortex hypofunction, specifically including inadequate cortisol production to meet the demands of significant physiological stress. This disease can be functionally grouped into three categories based on its underlying causes: primary, central, and hypothalamic-pituitary-adrenal (HPA) axis suppression (Table 1) [1,2]. Primary AI results from intrinsic pathology of both adrenal glands, leading to a deficiency in all adrenal hormones: cortisol, aldosterone, 19-carbon steroids, and secondarily also epinephrine [3,4]. Central AI, on the other hand, arises when cortisol synthesis is impaired due to inadequate pituitary adrenocorticotropic hormone (ACTH). This disruption is often due to the presence of tumors in the sella turcica or hypothalamic regions or can occur as a result of surgical or radiotherapy treatments targeting such tumors [5]. Central AI might be primarily a consequence of either pituitary or hypothalamic disease, or both, and it is neither possible nor practical to attempt to distinguish among these etiologies. HPA axis suppression is commonly induced after the sustained use of exogenous glucocorticoid medications and always involves hypothalamic corticotropin-releasing hormone (CRH) insufficiency. The important distinction is that HPA axis suppression is by definition reversible through a gradual discontinuation of the treatment, whereas primary and central AI are usually permanent [6].
- Lifesaving glucocorticoid medications have been widely available since the 1960s, and cortisol assays are routinely available throughout the world; nevertheless, significant challenges remain in both diagnosing AI and establishing the most efficacious replacement therapies. Within the context of this review, we attempt to synthesize important data from recent literature to present an efficient and reliable approach to the diagnosis of AI and to glucocorticoid replacement therapy for individuals with AI.
INTRODUCTION
- The presentation of AI is frequently non-specific, contributing to delays in diagnosis [7]. The manifestations of AI primarily resulting from cortisol deficiency include profound fatigue, loss of appetite, weight loss, nausea, vomiting, and low-grade fever. Additionally, individuals may experience body aches, joint pain, altered level of consciousness, and in primary AI, low blood pressure or orthostatic hypotension [1]. Androgen deficiency in women with AI leads to decreased body hair and reduced libido, as adrenal-derived androgens contribute significantly to the total androgen pool in women [8]. Conversely, in men, the primary source of androgens for body hair is the testes rather than the adrenals.
- Laboratory analyses in individuals with AI commonly show hyponatremia [9]. This physiological phenomenon arises from the insufficiency of cortisol, which leads to an inability to suppress arginine vasopressin from the hypothalamus. Consequently, cortisol deficiency impairs renal free water clearance, resulting in heightened water retention despite reduced plasma osmolality. These processes collectively underlie the euvolemic hyponatremia observed in individuals with all forms of AI [10]. In addition, aldosterone deficiency also contributes to hyponatremia in primary AI, particularly in young children, which leads to hypovolemic hyponatremia from renal loss of both water and sodium plus prerenal azotemia [11]. Additionally, laboratory investigations in both primary and central AI occasionally show normocytic anemia, eosinophilia, lymphopenia, and hypercalcemia [1].
- Nevertheless, while these symptoms might not be particularly informative, certain signs and symptoms can indirectly assist in determining the type of AI. For instance, in primary AI, concomitant aldosterone deficiency might manifest as salt cravings, postural hypotension, hyperkalemia, and metabolic acidosis [3]. Another noteworthy indicator is the presence of skin hyperpigmentation in primary AI. During a physical examination, healthcare providers may observe diffuse hyperpigmentation (bronzing) of the skin, which is often prominent in skin folds, the areolae, and the palmar creases. The pigmented deposition can also involve the oral mucosa, such as the palate, buccal area, and gums. Such alterations arise from very elevated ACTH production, which is characteristic of primary AI [12]. ACTH shares a structural resemblance to α-melanocyte stimulating hormone, which allows ACTH to activate the melanocortin type 1 receptor, which, in turn, stimulates melanin production in melanocytes [13].
IMPLICATIONS OF NON-SPECIFIC SIGNS AND SYMPTOMS IN PATIENTS WITH AI
- Basal hormones
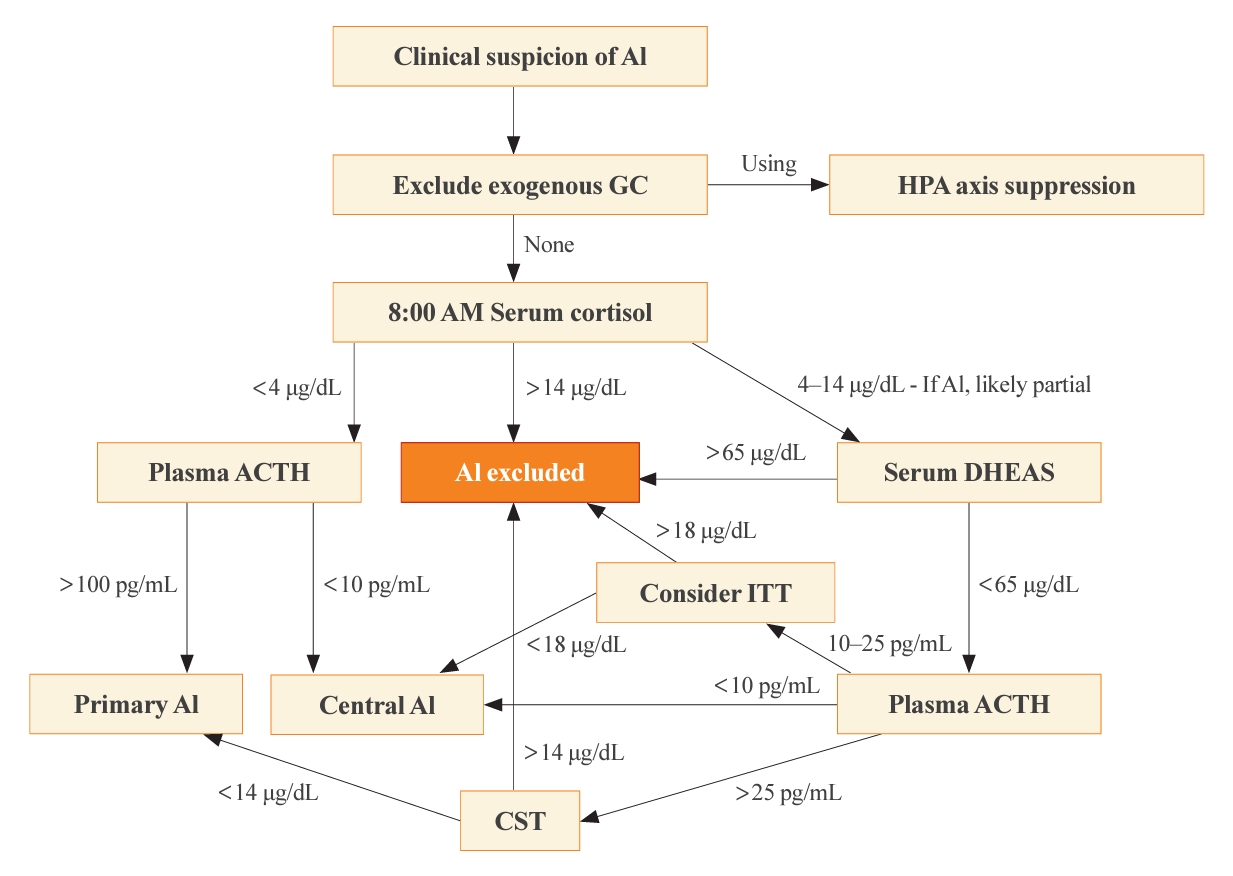
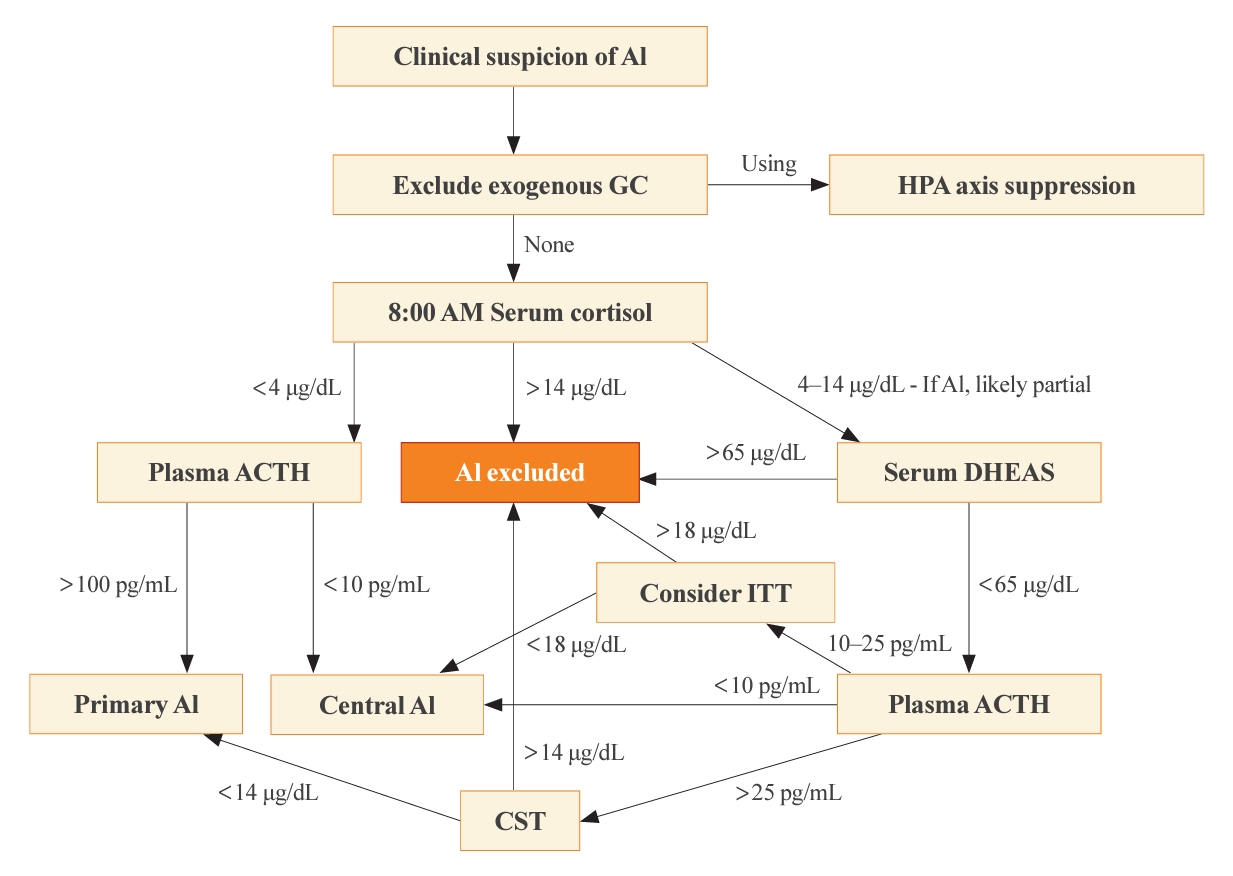
- The optimal time for measuring basal cortisol and ACTH is within a few hours after awakening, typically around 8:00 AM. This time encompasses the physiological peak of HPA axis activity [2]. In the appropriate clinical setting, a basal 8:00 AM serum cortisol of <4 µg/dL (110 nmol/L), combined with a plasma ACTH significantly exceeding the normal reference range (usually >100 pg/mL), establishes a diagnosis of primary AI without dynamic testing [4]. On the contrary, the diagnosis of central AI is often more challenging than primary AI, due to the absence of dynamic tests in most published studies of diagnostic criteria [1]. Generally, a morning serum cortisol of <4 µg/dL and the plasma ACTH below or near the low end of the normal range (typically <10 pg/mL), some degree of central AI is likely [5,14]. The presence of additional pituitary deficiencies, particularly central hypothyroidism, increases the likelihood of central AI. Conversely, morning serum cortisol of >14.5 µg/dL (>400 nmol/L) rules out significant dysfunction of the HPA axis. A single-center retrospective study revealed that basal serum cortisol >13 µg/dL (>350 nmol/L) measured by an immunoassay predicted normal adrenal function with a 100% negative predictive value [15]. This finding illustrates why basal morning serum cortisol testing serves as the initial step of screening for all forms of AI. A second basal serum test, dehydroepiandrosterone-sulfate (DHEAS), aids in the diagnosis of central AI when the 8:00 AM serum cortisol is 4 to 14 µg/dL [16]. A serum DHEAS level >65 µg/dL (>1,700 nmol/L) at any time of day in a patient not taking dehydroepiandrosterone (DHEA) supplements confirms normal HPA axis function without the need for dynamic testing (Fig. 1) [17]. One advantage of serum DHEAS is its limited circadian rhythm, which allows more frequent conclusive random testing in the afternoon, unlike cortisol. The main limitation of serum DHEAS is its normal decline with age, such that values <65 µg/dL in patients over 65 years old cannot be used to confirm AI.
- Cosyntropin stimulation test
- When the 8:00 AM serum cortisol is 4 to 14 µg/dL or when a diagnosis must be made later in the day for suspected primary AI, typically in sick hospitalized patients, a cosyntropin stimulation test (CST; tetracosactrin stimulation test) is the preferred dynamic test. This test involves the intravenous or intramuscular injection of 250 µg cosyntropin (ACTH1-24), a synthetic fragment of ACTH with full biological activity, and subsequent serum cortisol measurements 30 to 60 minutes later. Although a 1-µg CST has been proposed as a better test for central AI, we do not recommend this test for diagnosis of primary AI, although lower doses might be used to diagnose primary AI when cosyntropin is in short supply [4].
- Previously, criteria for the diagnosis of primary AI required that serum cortisol remain <18 µg/dL (<500 nmol/L) at both 30 and 60 minutes after cosyntropin [4,5]. Older literature included a rise of >9 µg/dL (>250 nmol/L) from baseline [18], but this criterion has been abandoned, because the magnitude of the rise merely reflects the time of day and thus baseline cortisol value. It is important to recognize that these criteria derive from studies of normal, healthy volunteers and represent the low end of this normative group, rather than the upper end of patients with proven primary AI. In addition, a cosyntropin dose of 250 µg is grossly supraphysiologic, and the normative data do not imply that circulating cortisol concentrations of <18 µg/dL are insufficient to survive major physiological stress events. Furthermore, the polyclonal immunoassays on which these criteria were based have been replaced with more specific monoclonal immunoassays and mass spectrometry measurements, which yield cortisol values 20% to 30% lower than the prior assays [19]. A retrospective analysis of CST results performed in patients suspected of AI recommended that threshold 30-minute post-stimulation cortisol values of 14.6 µg/dL for Elecsys II (Roche Diagnostics GmbH, Mannheim, Germany) and 14.8 µg/dL for Access platform immunoassays (Beckman Coulter, Inc., Brea, CA, USA), to reduce false positives [20]. Liquid chromatography with tandem mass spectrometry (LC-MS/MS) is now considered the gold standard method to measure serum steroids; however, LC-MS/MS is not widely available, especially for same-day results, although threshold values are similar for the monoclonal immunoassays. Even these criteria for modern assays carry the same caveats as discussed for the older assays.
- In cases of central AI or HPA axis suppression, the loss of adrenal cortisol response following ACTH deficiency develops gradually over time. Consequently, it is not advisable to employ the CST during the early phases of central AI to avoid falsely normal responses, particularly within the initial 6 weeks following a pituitary insult [21]. Additionally, some centers have used synthetic CRH to evaluate the function of the HPA axis; however, CRH testing has not been shown to be superior to CST, and CRH is no longer commercially available [22]. In interpreting basal and dynamic serum cortisol results, it is imperative to determine if exogenous glucocorticoids that interfere with the cortisol assay, such as hydrocortisone or prednisolone, have been administered recently. These medications must be withheld for at least 24 hours prior to testing, as hydrocortisone is cortisol, and prednisolone still cross-reacts in monoclonal immunoassays but not LC-MS/MS cortisol assays [23].
- Other tests for diagnosis of central AI and HPA axis suppression
- Historically, the insulin tolerance test (ITT) has been widely regarded as the gold standard for diagnosing all forms of AI, particularly those involving hypothalamic and pituitary dysfunction, in which the CST can give a false-negative normal cortisol rise. By administering a bolus of intravenous insulin (0.1 unit/kg) and inducing severe hypoglycemia, the counter-regulatory response activates and thus interrogates the entire HPA axis. The glucose nadir is typically 30 to 45 minutes after the insulin bolus, and the criterion for central AI is a peak serum cortisol <18 µg/dL (<500 nmol/L) at 60 to 90 minutes [24]. The ITT, however, entails significant risks to the patients, necessitates a high degree of supervision, and is contraindicated among patients with a history of cardiovascular disease or seizures. Furthermore, threshold values for ITT have not been established for modern cortisol assays. As a result, the ITT has become disfavored in contemporary clinical practice and is rarely necessary after basal cortisol and DHEAS testing (Fig. 1).
- The metyrapone test serves as an alternative to ITT for assessing the function of the entire HPA axis [25]. Metyrapone is an inhibitor of the 11β-hydroxylase enzyme, and a 30 mg/kg oral dose (up to 3 g) given with food at 11:00 PM will prevent the normal early morning cortisol rise. Loss of negative feedback enhances ACTH secretion, and steroid precursors accumulate upstream of the pharmacological blockade. Blood samples are collected 8 hours after metyrapone administration for plasma ACTH, serum 11-deoxycortisol, and serum cortisol. A serum cortisol <5 µg/dL is required to confirm adequate 11β-hydroxylase inhibition and to validate the test. If the serum cortisol is low, an increase in plasma 11-deoxycortisol to >7 µg/dL (>200 nmol/L) is indicative of an intact HPA axis [26]. Certain research studies suggest using the criterion sum of serum 11-deoxycortisol plus cortisol >16 µg/dL (>450 nmol/L) to mitigate cross-reactivity of 11-deoxycortisol in cortisol assays [27]. Conceptually, the threshold is similar to the ITT criterion of >18 µg/dL (>500 nmol/L). Moreover, plasma ACTH measurement (<3-fold rise) makes the metyrapone test more sensitive than steroid measurements alone for detecting central AI [26]. The limited availability of metyrapone in numerous countries, high cost, and the substantial heterogeneity observed in 11-deoxycortisol assays, however, collectively restrict the widespread use of metyrapone testing, which can also precipitate an adrenal crisis in a symptomatic and ill patient.
- The glucagon stimulation test (GST) has been used widely to diagnose growth hormone deficiency and can be a useful diagnostic test for central AI. A 1 mg intravenous glucagon bolus raises glucose transiently and provokes insulin secretion in non-diabetic patients. This insulin pulse lowers glucose and precipitates a counter-regulatory response during the 240 minutes after the bolus, including a rise in cortisol. The value of the GST is unclear, however, due to differing proposed thresholds for peak post-stimulation cortisol concentrations, which has led to variable reports of sensitivity and specificity [28,29]. Furthermore, a prospective comparison between GST and the metyrapone test in patients with suspected ACTH deficiency, using a peak cortisol threshold of ≥16 µg/dL (≥440 nmol/L), revealed a significant discrepancy between the two tests in the proportion of patients diagnosed with AI [30], which underscores the need for caution when employing the GST for assessing ACTH reserve.
- Noninvasive diagnostic testing
- Basal and stimulated morning salivary cortisol values have been used as an approach to the diagnosis of AI. A prospective study employing an enzyme-linked immunoassay kit demonstrated that the performance of cosyntropin-stimulated salivary and serum cortisol were comparable for AI diagnosis. The optimal cutoff value to exclude AI for stimulated salivary cortisol in this study was >470 ng/dL (>13 nmol/L) [31]. Another retrospective study using LC-MS/MS to assess basal salivary cortisol found that a threshold of <30 ng/dL (<1.0 nmol/L) could be used to confirm AI with a positive predictive value of 100% [32].
- The salivary gland epithelial cells express the 11β-hydroxysteroid dehydrogenase type 2 enzyme, which oxidizes cortisol to cortisone. Consequently, the concentration of cortisone is typically four to six times higher than that of cortisol in saliva. A prospective study of both salivary cortisol and cortisone measured by LC-MS/MS after cosyntropin stimulation demonstrated that salivary cortisone can be used as a diagnostic test for AI [33]. Furthermore, a recent prospective study using waking salivary cortisone in a non-hospital setting provided data regarding the accuracy of AI diagnosis. A waking salivary cortisone <250 ng/dL (<7 nmol/L) served as a cutoff to confirm AI, whereas values >600 ng/dL (>17 nmol/L) excluded AI [34]. Moreover, the measurement of salivary cortisol or cortisone 60 minutes after nasal administration of Nasacthin003 (patent pending), a 500 µg cosyntropin dose mixed with mucoadhesive chitosan, presents a completely noninvasive alternative test for AI compared to the conventional CST [35]. These salivary tests hold considerable promise, particularly in children, owing to their simplicity, non-invasiveness, and ease of administration. Additionally, salivary cortisol and cortisone have an advantage over serum cortisol measurements because they are not influenced by binding proteins like albumin or corticosteroid-binding globulin.
LABORATORY INVESTIGATIONS FOR AI DIAGNOSIS
- The objectives of long-term AI treatment are to achieve the optimal glucocorticoid (and mineralocorticoid in primary AI) replacement regimen, to maintain quality of life, to avoid the adverse consequences of excessive replacement, and to prevent mortality, particularly from adrenal crises, thorough education and preparation [1,2].
- Glucocorticoid replacement
- Endogenous cortisol secretion exhibits a circadian pattern, peaking shortly after awakening and gradually declining over the course of the day, with a slight increase at mealtimes [36]. In healthy individuals, the adrenal glands produce approximately 5 to 10 mg of cortisol per square meter of body surface area daily [37]. This production rate equates to a recommended daily oral replacement dose of 10 to 20 mg of hydrocortisone [38]. Generally, patients with primary AI require a complete replacement of their daily cortisol production, whereas those with central AI often require a lower replacement dose, as they may retain some residual ACTH and cortisol secretion [1]. Hydrocortisone is typically the preferred medication, while cortisone acetate has fallen out of favor due to its reliance on hepatic activation for conversion to cortisol, which leads to a slightly delayed onset of action [38]. Because of its short half-life (60 to 90 minutes), hydrocortisone is typically administered two or three times daily, with the highest dose provided in the morning immediately upon awakening. For a two-dose regimen, the next dose is administered in the early afternoon (around 2:00 PM), while for a three-dose regimen, the latter two doses are given at lunch and early evening but not within 4 hours of bedtime. These dosing regimens aim to approximate the natural circadian rhythm of cortisol as closely as possible, with the final dose scheduled approximately 6 hours before bedtime to prevent sleep disturbances. In some countries, prednisolone remains the only treatment option for AI despite concerns regarding the more frequent occurrence of dyslipidemia [39] and reduced bone mineral density [40] in comparison to hydrocortisone. Prescribed in doses ranging from 3 to 5 mg/day, prednisolone, however, is cost-effective and can be administered once daily, making it a potential alternative to hydrocortisone, particularly in patients with difficulty taking midday doses [4]. Dexamethasone, on the other hand, is not recommended for AI treatment due to its high potency, lack of tablet sizes in the physiological window, and long half-life, which often result in over-treatment and Cushingoid side effects [38].
- Stress dosing
- Individualized prescriptions of supplementary glucocorticoids are crucial during times of physiological stress to prevent adrenal crises [41]. To replicate the HPA axis stress response, the usual approach for oral hydrocortisone stress dosing is to double or triple the regular replacement dose, depending on the severity of the stressor, sometimes with a more even dose distribution throughout the day. However, pharmacokinetics of oral hydrocortisone varies significantly among individuals, with up to a 10-fold difference in drug metabolism rates. Fast metabolizers, characterized by a high clearance rate of hydrocortisone, might not achieve sufficient drug exposure by simply doubling the hydrocortisone dose [42]. This variability necessitates personalized adjustments in treatment doses. Additionally, patients who are unable to take oral glucocorticoids due to gastrointestinal illness or major surgery require parenteral administration. The conventional outpatient rescue approach is to administer 100 mg of hydrocortisone hemisuccinate intramuscularly, which is rapidly reconstituted in a partitioned vial (Solu-Cortef Act-O-Vial, Pfizer Inc., New York, NY, USA). This single dose provides adequate cortisol replacement for at least 4 hours, during which time the patient should be promptly transported to a medical facility, by ambulance if necessary, for intravenous fluid resuscitation and subsequent hydrocortisone doses. Although an additional 100 mg dose every 8 hours for every patient with AI who is hospitalized has been taught to internists for decades, this dose is excess, and smaller, more frequent doses are preferable, such as 25 to 50 mg every 6 hours. Based on current evidence, hydrocortisone doses should not exceed 200 mg/day, no matter how severe the illness [43]. Conversely, patients should not routinely conduct stress doses for periods of mental stress or minor illnesses such as afebrile sore throat or rhinorrhea. Such practice leads to unnecessary glucocorticoid exposure and its consequences.
- Alternative therapies in development
- Given the awkward dosing schedules required to mimic the circadian cortisol rhythm, alternative strategies have been studied, such as modified-release hydrocortisone formulations [2]. Plenadren (Takeda Pharmaceuticals International AG Ireland Branch, Dublin, Ireland), the first modified-release hydrocortisone, features a dual-release kinetics system consisting of a coated outer layer, which provides an immediate-release of hydrocortisone, and an extended-release inner core [44]. This formulation technology allows once-daily administration and better mimics the physiological cortisol rhythm throughout the circadian rhythm than conventional immediate-release hydrocortisone [36]. There have been reports of favorable metabolic effects, including impacts on weight, blood pressure, glucose levels, lipid profiles, and immune cell profiles, in patients using Plenadren [44-47].
- Chronocort (Diurnal Ltd., a Neurocrine Biosciences Company, San Diego, CA, USA), another modified-release hydrocortisone formulation, differs from Plenadren by having a single delayed and sustained absorption profile, rather than a dual-release mechanism [48]. The Chronocort system involves capsulated granules, comprising an inert core onto which hydrocortisone is coated, covered with a pH-sensitive delayed-release layer. In trials for 21-hydroxylase deficiency (21OHD), the initial Chronocort regimen consisted of a 20 mg dose at approximately 11:00 PM, followed by a smaller 10 mg dose at approximately 7:00 AM. The higher nighttime dose allows the gradual release of hydrocortisone during the early morning hours, effectively addressing the overnight increase in ACTH and adrenal androgens and simultaneously mimicking the circadian profile. Meanwhile, the morning dose provides adequate glucocorticoid coverage throughout the day but might not be necessary in patients with AI not due to 21OHD [36]. In patients with 21OHD, Chronocort treatment twice-daily reduced 17-hydroxyprogesterone and androstenedione biomarkers better than immediate-release hydrocortisone thrice daily and allowed for a reduction in the total daily glucocorticoid dose [49,50].
- Continuous subcutaneous hydrocortisone infusion (CSHI) using a programmable infusion pump offers a theoretically ideal alternative for glucocorticoid replacement to best replicate the natural cortisol circadian rhythm [51]. This approach can be particularly beneficial for individuals with atypical hydrocortisone pharmacokinetics, such as reduced hydrocortisone bioavailability or increased hydrocortisone clearance [52]. The same hydrocortisone hemisuccinate (Solu-Cortef) used for rescue intramuscular injection is used in the pump, but this preparation has not been formulated, tested, or approved by regulatory agencies for this purpose.
- Whether CSHI enhances the overall quality of life for patients with AI; however, remains controversial. In addition to the risk of pump malfunction, the limitations of CSHI include high cost, invasiveness compared to oral therapy, and possible skin reactions at the catheter site [53].
- Mineralocorticoid replacement
- Patients with primary AI, as well as individuals who have undergone bilateral adrenalectomy, generally benefit from mineralocorticoid replacement; however, up to 13.5% of individuals with autoimmune Addison disease may retain some residual aldosterone production, particularly early after diagnosis [54]. The aims of mineralocorticoid therapy in primary AI are to maintain volume status, normal blood pressure, electrolyte balance, and to mitigate salt cravings [1]. Insufficient replacement of mineralocorticoids may be responsible for unfavorable cardiometabolic outcomes and a diminished overall sense of well-being in individuals with primary AI [55].
- Because aldosterone is expensive to manufacture and has poor pharmacokinetics, patients with primary AI are treated with fludrocortisone, typically 0.05 to 0.20 mg in a daily morning dose [2]. This timing aligns with the natural circadian rhythm of aldosterone secretion, which peaks at 8:00 AM and nadirs at 11:00 PM, similar to cortisol [56]. Some patients empirically require higher or twice-daily fludrocortisone dosing [1], and patients treated with prednisolone or dexamethasone as glucocorticoid replacement often require high fludrocortisone doses, owing to their negligible mineralocorticoid activity relative to hydrocortisone. Infants and young children require proportionately higher fludrocortisone doses than most adults, as the sensitivity of the newborn kidney to mineralocorticoids is lower than in adults. Consequently, the absolute dose of fludrocortisone is similar throughout life and does not increase linearly with body surface area, as does the dose of glucocorticoid [1]. Temporary 50% to 100% increments of the usual fludrocortisone dose may be recommended in hot climates and conditions that promote excessive sweating, such as vigorous and long-lasting exercise programs [55]. Patients with mineralocorticoid deficiency are typically allowed to consume sodium in the diet and salty foods without restriction. Medications that affect blood pressure and electrolytes, including diuretics, might necessitate fludrocortisone dose adjustments and should be avoided [1].
- Fludrocortisone therapy is primarily titrated based on clinical evaluation of volume status. The effects of fludrocortisone in regulating extracellular fluid volume and blood pressure are chronic and gradual. Consequently, clinical improvement will not be manifest immediately following a dosage change and should not be reassessed for a minimum of 2 to 4 weeks afterward [57]. Sitting versus standing blood pressure and heart rate, serum potassium, salt craving, and plasma renin (mass or activity) are used for titrating mineralocorticoid therapy for primary AI. Good control is evidenced with no orthostatic blood pressure drop or heart rate increase, normal serum potassium, mild salt craving, and plasma renin below the upper limit of the normal range. These parameters do not always change in parallel, so clinical judgment is necessary. For example, hypokalemia may occur with plasma renin in the normal range during fludrocortisone treatment [58].
- Patients with primary AI who develop hypertension during treatment should have their hydrocortisone dose re-evaluated and the fludrocortisone dose reduced if plasma renin and/or serum potassium are low but generally not discontinued. Antihypertensive drugs other than diuretics, such as calcium channel blockers, might be added in euvolemic AI patients [1].
- Patients with primary AI and fatigue that does not improve within 2 hours after a dose of hydrocortisone are typically volume depleted from fludrocortisone under-treatment, not inadequate glucocorticoid dosing.
- Adrenal androgen (DHEA) replacement
- Adrenal androgen deficiency occurs in both primary and central AI, which leads to reduced androgen-dependent hair and possibly sexual interest in women. Adrenal androgen replacement with DHEA at a daily dose ranging from 10 to 25 mg might be beneficial in improving libido and mental well-being, both via conversion to testosterone and possibly through direct effects as well [8,59]. In addition to androgen-related side effects such as hirsutism, acne, and oily skin, DHEA can also be converted into estrogen, which presents a currently unquantified risk of estrogen-sensitive cancers, cardiovascular effects, and venous thrombosis. Long-term safety data for DHEA are limited, and Endocrine Society clinical practice guidelines do not recommend its routine use [4,60].
REPLACEMENT THERAPY FOR AI
- Prompt diagnosis of AI is of utmost importance, to avert life-threatening conditions during stressful situations when the cortisol response is compromised. In parallel, an incorrect diagnosis of AI subjects a patient to a lifetime of glucocorticoid therapy and should be avoided, particularly when reversible HPA axis suppression from exogenous glucocorticoids lowers serum cortisol below certain thresholds. Despite the availability of diagnostic techniques for decades, the best strategy and threshold values for confirming or ruling out AI are not known. Efforts have been made to enhance diagnostic accuracy through the development of simplified techniques and improved assays. Standardized studies that include large numbers of patients with established AI are necessary to establish standardized cutoff values. Furthermore, even after diagnosis, challenges persist in replicating the natural cortisol and mineralocorticoid rhythms. Well-designed, double-blind, randomized studies comparing various medication regimens and formulations are still needed to address these ongoing dilemmas.
CONCLUSIONS
-
CONFLICTS OF INTEREST
Richard J. Auchus reports consulting fees and contracted research support from Diurnal, Ltd. and Neurocrine Biosciences. Suranut Charoensri has nothing to declare.
Article information
- 1. Hahner S, Ross RJ, Arlt W, Bancos I, Burger-Stritt S, Torpy DJ, et al. Adrenal insufficiency. Nat Rev Dis Primers 2021;7:19.ArticlePubMedPDF
- 2. Husebye ES, Pearce SH, Krone NP, Kampe O. Adrenal insufficiency. Lancet 2021;397:613–29.ArticlePubMed
- 3. Barthel A, Benker G, Berens K, Diederich S, Manfras B, Gruber M, et al. An update on Addison’s disease. Exp Clin Endocrinol Diabetes 2019;127:165–75.ArticlePubMed
- 4. Bornstein SR, Allolio B, Arlt W, Barthel A, Don-Wauchope A, Hammer GD, et al. Diagnosis and treatment of primary adrenal insufficiency: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 2016;101:364–89.ArticlePubMedPMC
- 5. Fleseriu M, Hashim IA, Karavitaki N, Melmed S, Murad MH, Salvatori R, et al. Hormonal replacement in hypopituitarism in adults: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 2016;101:3888–921.ArticlePubMed
- 6. Prete A, Bancos I. Glucocorticoid induced adrenal insufficiency. BMJ 2021;374:n1380.ArticlePubMed
- 7. Papierska L, Rabijewski M. Delay in diagnosis of adrenal insufficiency is a frequent cause of adrenal crisis. Int J Endocrinol 2013;2013:482370.ArticlePubMedPMCPDF
- 8. Arlt W, Callies F, van Vlijmen JC, Koehler I, Reincke M, Bidlingmaier M, et al. Dehydroepiandrosterone replacement in women with adrenal insufficiency. N Engl J Med 1999;341:1013–20.ArticlePubMed
- 9. Diederich S, Franzen NF, Bahr V, Oelkers W. Severe hyponatremia due to hypopituitarism with adrenal insufficiency: report on 28 cases. Eur J Endocrinol 2003;148:609–17.ArticlePubMed
- 10. Garrahy A, Thompson CJ. Hyponatremia and glucocorticoid deficiency. Front Horm Res 2019;52:80–92.ArticlePubMed
- 11. Benner BJ, Alsma J, Feelders RA. Hyponatraemia and hyperpigmentation in primary adrenal insufficiency. BMJ Case Rep 2019;12:e227200.ArticlePubMedPMC
- 12. Mohamed F, Raal FJ. Hyperpigmentation from Addison’s disease. N Engl J Med 2021;384:1752.ArticlePubMed
- 13. Abdel-Malek ZA, Knittel J, Kadekaro AL, Swope VB, Starner R. The melanocortin 1 receptor and the UV response of human melanocytes: a shift in paradigm. Photochem Photobiol 2008;84:501–8.ArticlePubMed
- 14. Grossman AB. Clinical review: the diagnosis and management of central hypoadrenalism. J Clin Endocrinol Metab 2010;95:4855–63.PubMed
- 15. Kumar R, Carr P, Wassif W. Diagnostic performance of morning serum cortisol as an alternative to short synacthen test for the assessment of adrenal reserve; a retrospective study. Postgrad Med J 2022;98:113–8.ArticlePubMedPDF
- 16. Charoensri S, Chailurkit L, Muntham D, Bunnag P. Serum dehydroepiandrosterone sulfate in assessing the integrity of the hypothalamic-pituitary-adrenal axis. J Clin Transl Endocrinol 2017;7:42–6.ArticlePubMedPMC
- 17. Nasrallah MP, Arafah BM. The value of dehydroepiandrosterone sulfate measurements in the assessment of adrenal function. J Clin Endocrinol Metab 2003;88:5293–8.ArticlePubMed
- 18. Rothwell PM, Udwadia ZF, Lawler PG. Cortisol response to corticotropin and survival in septic shock. Lancet 1991;337:582–3.ArticlePubMed
- 19. Brossaud J, Gatta B, Tabarin A, Corcuff JB. Different methods to estimate serum free cortisol: a comparison during cortisol tetracosactide testing. Clin Chem Lab Med 2015;53:1367–73.ArticlePubMed
- 20. Javorsky BR, Raff H, Carroll TB, Algeciras-Schimnich A, Singh RJ, Colon-Franco JM, et al. New cutoffs for the biochemical diagnosis of adrenal insufficiency after ACTH stimulation using specific cortisol assays. J Endocr Soc 2021;5:bvab022.ArticlePubMedPMCPDF
- 21. Ospina NS, Al Nofal A, Bancos I, Javed A, Benkhadra K, Kapoor E, et al. ACTH stimulation tests for the diagnosis of adrenal insufficiency: systematic review and meta-analysis. J Clin Endocrinol Metab 2016;101:427–34.ArticlePubMed
- 22. Schmidt IL, Lahner H, Mann K, Petersenn S. Diagnosis of adrenal insufficiency: evaluation of the corticotropin-releasing hormone test and Basal serum cortisol in comparison to the insulin tolerance test in patients with hypothalamic-pituitary-adrenal disease. J Clin Endocrinol Metab 2003;88:4193–8.ArticlePubMed
- 23. Krasowski MD, Drees D, Morris CS, Maakestad J, Blau JL, Ekins S. Cross-reactivity of steroid hormone immunoassays: clinical significance and two-dimensional molecular similarity prediction. BMC Clin Pathol 2014;14:33.ArticlePubMedPMCPDF
- 24. Erturk E, Jaffe CA, Barkan AL. Evaluation of the integrity of the hypothalamic-pituitary-adrenal axis by insulin hypoglycemia test. J Clin Endocrinol Metab 1998;83:2350–4.ArticlePubMed
- 25. Giordano R, Picu A, Bonelli L, Balbo M, Berardelli R, Marinazzo E, et al. Hypothalamus-pituitary-adrenal axis evaluation in patients with hypothalamo-pituitary disorders: comparison of different provocative tests. Clin Endocrinol (Oxf) 2008;68:935–41.ArticlePubMed
- 26. Steiner H, Bahr V, Exner P, Oelkers PW. Pituitary function tests: comparison of ACTH and 11-deoxy-cortisol responses in the metyrapone test and with the insulin hypoglycemia test. Exp Clin Endocrinol 1994;102:33–8.ArticlePubMed
- 27. Berneis K, Staub JJ, Gessler A, Meier C, Girard J, Muller B. Combined stimulation of adrenocorticotropin and compound-S by single dose metyrapone test as an outpatient procedure to assess hypothalamic-pituitary-adrenal function. J Clin Endocrinol Metab 2002;87:5470–5.ArticlePubMed
- 28. Ach T, Yosra H, Jihen M, Abdelkarim Asma B, Maha K, Molka C, et al. Cortisol cut-points for the glucagon stimulation test in the evaluation of hypothalamic pituitary adrenal axis. Endocr J 2018;65:935–42.ArticlePubMed
- 29. Hamrahian AH, Yuen KC, Gordon MB, Pulaski-Liebert KJ, Bena J, Biller BM. Revised GH and cortisol cut-points for the glucagon stimulation test in the evaluation of GH and hypothalamic-pituitary-adrenal axes in adults: results from a prospective randomized multicenter study. Pituitary 2016;19:332–41.ArticlePubMedPDF
- 30. Cegla J, Jones B, Seyani L, Papadoulou D, Wynne K, Martin NM, et al. Comparison of the overnight metyrapone and glucagon stimulation tests in the assessment of secondary hypoadrenalism. Clin Endocrinol (Oxf) 2013;78:738–42.ArticlePubMed
- 31. Kim YJ, Kim JH, Hong AR, Park KS, Kim SW, Shin CS, et al. Stimulated salivary cortisol as a noninvasive diagnostic tool for adrenal insufficiency. Endocrinol Metab (Seoul) 2020;35:628–35.ArticlePubMedPMCPDF
- 32. Langelaan ML, Kisters JM, Oosterwerff MM, Boer AK. Salivary cortisol in the diagnosis of adrenal insufficiency: cost efficient and patient friendly. Endocr Connect 2018;7:560–6.ArticlePubMedPMC
- 33. Mak IY, Au Yeung BY, Ng YW, Choi CH, Iu HY, Shek CC, et al. Salivary cortisol and cortisone after low-dose corticotropin stimulation in the diagnosis of adrenal insufficiency. J Endocr Soc 2017;1:96–108.ArticlePubMedPMC
- 34. Debono M, Caunt S, Elder C, Fearnside J, Lewis J, Keevil B, et al. Real world evidence supports waking salivary cortisone as a screening test for adrenal insufficiency. Clin Endocrinol (Oxf) 2023;99:517–24.ArticlePubMed
- 35. Elder CJ, Vilela R, Johnson TN, Taylor RN, Kemp EH, Keevil BG, et al. Pharmacodynamic studies of nasal tetracosactide with salivary glucocorticoids for a noninvasive Short Synacthen Test. J Clin Endocrinol Metab 2020;105:dgaa323.ArticlePubMedPDF
- 36. Debono M, Ghobadi C, Rostami-Hodjegan A, Huatan H, Campbell MJ, Newell-Price J, et al. Modified-release hydrocortisone to provide circadian cortisol profiles. J Clin Endocrinol Metab 2009;94:1548–54.ArticlePubMedPMCPDF
- 37. Esteban NV, Yergey AL. Cortisol production rates measured by liquid chromatography/mass spectrometry. Steroids 1990;55:152–8.ArticlePubMed
- 38. Kumar R, Wassif WS. Adrenal insufficiency. J Clin Pathol 2022;75:435–42.ArticlePubMed
- 39. Quinkler M, Ekman B, Marelli C, Uddin S, Zelissen P, Murray RD, et al. Prednisolone is associated with a worse lipid profile than hydrocortisone in patients with adrenal insufficiency. Endocr Connect 2017;6:1–8.ArticlePubMedPMC
- 40. Frey KR, Kienitz T, Schulz J, Ventz M, Zopf K, Quinkler M. Prednisolone is associated with a worse bone mineral density in primary adrenal insufficiency. Endocr Connect 2018;7:811–8.ArticlePubMedPMC
- 41. Rushworth RL, Torpy DJ, Falhammar H. Adrenal crisis. N Engl J Med 2019;381:852–61.ArticlePubMed
- 42. Werumeus Buning J, Touw DJ, Brummelman P, Dullaart RP, van den Berg G, van der Klauw MM, et al. Pharmacokinetics of oral hydrocortisone: results and implications from a randomized controlled trial. Metabolism 2017;71:7–16.ArticlePubMed
- 43. Arafah BM. Hypothalamic pituitary adrenal function during critical illness: limitations of current assessment methods. J Clin Endocrinol Metab 2006;91:3725–45.ArticlePubMedPDF
- 44. Johannsson G, Nilsson AG, Bergthorsdottir R, Burman P, Dahlqvist P, Ekman B, et al. Improved cortisol exposure-time profile and outcome in patients with adrenal insufficiency: a prospective randomized trial of a novel hydrocortisone dual-release formulation. J Clin Endocrinol Metab 2012;97:473–81.PubMed
- 45. Isidori AM, Venneri MA, Graziadio C, Simeoli C, Fiore D, Hasenmajer V, et al. Effect of once-daily, modified-release hydrocortisone versus standard glucocorticoid therapy on metabolism and innate immunity in patients with adrenal insufficiency (DREAM): a single-blind, randomised controlled trial. Lancet Diabetes Endocrinol 2018;6:173–85.ArticlePubMed
- 46. Nilsson AG, Bergthorsdottir R, Burman P, Dahlqvist P, Ekman B, Engstrom BE, et al. Long-term safety of once-daily, dual-release hydrocortisone in patients with adrenal insufficiency: a phase 3b, open-label, extension study. Eur J Endocrinol 2017;176:715–25.ArticlePubMedPMC
- 47. Quinkler M, Miodini Nilsen R, Zopf K, Ventz M, Oksnes M. Modified-release hydrocortisone decreases BMI and HbA1c in patients with primary and secondary adrenal insufficiency. Eur J Endocrinol 2015;172:619–26.ArticlePubMed
- 48. Whitaker M, Debono M, Huatan H, Merke D, Arlt W, Ross RJ. An oral multiparticulate, modified-release, hydrocortisone replacement therapy that provides physiological cortisol exposure. Clin Endocrinol (Oxf) 2014;80:554–61.ArticlePubMedPMC
- 49. Mallappa A, Sinaii N, Kumar P, Whitaker MJ, Daley LA, Digweed D, et al. A phase 2 study of Chronocort, a modified-release formulation of hydrocortisone, in the treatment of adults with classic congenital adrenal hyperplasia. J Clin Endocrinol Metab 2015;100:1137–45.ArticlePubMedPMC
- 50. Jones CM, Mallappa A, Reisch N, Nikolaou N, Krone N, Hughes BA, et al. Modified-release and conventional glucocorticoids and diurnal androgen excretion in congenital adrenal hyperplasia. J Clin Endocrinol Metab 2017;102:1797–806.ArticlePubMedPMC
- 51. Oksnes M, Bjornsdottir S, Isaksson M, Methlie P, Carlsen S, Nilsen RM, et al. Continuous subcutaneous hydrocortisone infusion versus oral hydrocortisone replacement for treatment of Addison’s disease: a randomized clinical trial. J Clin Endocrinol Metab 2014;99:1665–74.ArticlePubMed
- 52. Hindmarsh PC. The child with difficult to control congenital adrenal hyperplasia: is there a place for continuous subcutaneous hydrocortisone therapy. Clin Endocrinol (Oxf) 2014;81:15–8.PubMed
- 53. Mallappa A, Nella AA, Sinaii N, Rao H, Gounden V, Perritt AF, et al. Long-term use of continuous subcutaneous hydrocortisone infusion therapy in patients with congenital adrenal hyperplasia. Clin Endocrinol (Oxf) 2018;89:399–407.ArticlePubMedPMCPDF
- 54. Sævik AB, Akerman AK, Methlie P, Quinkler M, Jorgensen AP, Hoybye C, et al. Residual corticosteroid production in autoimmune Addison disease. J Clin Endocrinol Metab 2020;105:2430–41.ArticlePubMedPMCPDF
- 55. Esposito D, Pasquali D, Johannsson G. Primary adrenal insufficiency: managing mineralocorticoid replacement therapy. J Clin Endocrinol Metab 2018;103:376–87.ArticlePubMed
- 56. Williams GH, Cain JP, Dluhy RG, Underwood RH. Studies of the control of plasma aldosterone concentration in normal man. I. Response to posture, acute and chronic volume depletion, and sodium loading. J Clin Invest 1972;51:1731–42.ArticlePubMedPMC
- 57. Smith SJ, MacGregor GA, Markandu ND, Bayliss J, Banks RA, Prentice MG, et al. Evidence that patients with Addison’s disease are undertreated with fludrocortisone. Lancet 1984;1:11–4.ArticlePubMed
- 58. Inder WJ, Meyer C, Hunt PJ. Management of hypertension and heart failure in patients with Addison’s disease. Clin Endocrinol (Oxf) 2015;82:789–92.ArticlePubMed
- 59. Alkatib AA, Cosma M, Elamin MB, Erickson D, Swiglo BA, Erwin PJ, et al. A systematic review and meta-analysis of randomized placebo-controlled trials of DHEA treatment effects on quality of life in women with adrenal insufficiency. J Clin Endocrinol Metab 2009;94:3676–81.ArticlePubMed
- 60. Wierman ME, Arlt W, Basson R, Davis SR, Miller KK, Murad MH, et al. Androgen therapy in women: a reappraisal: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2014;99:3489–510.ArticlePubMed
 PubReader
PubReader ePub Link
ePub Link Cite
Cite