
Articles
- Page Path
- HOME > Endocrinol Metab > Volume 29(1); 2014 > Article
-
Case ReportPheochromocytoma with Markedly Abnormal Liver Function Tests and Severe Leukocytosis
- Chai Ryoung Eun, Jae Hee Ahn, Ji A Seo, Nan Hee Kim
-
Endocrinology and Metabolism 2014;29(1):83-90.
DOI: https://doi.org/10.3803/EnM.2014.29.1.83
Published online: March 14, 2014
Division of Endocrinology and Metabolism, Department of Internal Medicine, Korea University College of Medicine, Seoul, Korea.
- Corresponding author: Nan Hee Kim. Division of Endocrinology and Metabolism, Department of Internal Medicine, Korea University Ansan Hospital, Korea University College of Medicine, 123 Jeokgeum-ro, Danwon-gu, Ansan 425-707, Korea. Tel: +82-31-412-5952, Fax: +82-31-412-5984, nhkendo@gmail.com
• Received: January 15, 2013 • Accepted: March 21, 2013
Copyright © 2014 Korean Endocrine Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
- Pheochromocytoma is a rare neuroendocrine tumor arising from the medulla of the adrenal glands, which causes an overproduction of catecholamines. The common symptoms are headache, palpitations, and sweating; however, various other clinical manifestations might also be present. Accurate diagnosis of pheochromocytoma is important because surgical treatment is usually successful, and associated clinical problems are reversible if treated early. A 49-year-old man with a history of uncontrolled hypertension and diabetes mellitus presented with chest pain, fever, and sweating. His liver function tests and white blood cell counts were markedly increased and his echocardiography results suggested stress-induced cardiomyopathy. His abdominal computed tomography showed a 5×5-cm-sized tumor in the left adrenal gland, and laboratory tests confirmed catecholamine overproduction. After surgical resection of the left adrenal gland, his liver function tests and white blood cell counts normalized, and echocardiography showed normal cardiac function. Moreover, his previous antihypertensive regimen was deescalated, and his previously uncontrolled blood glucose levels normalized without medication.
- Pheochromocytoma is a rare tumor arising from chromaffin cells of the sympathetic nervous system which releases catecholamines. The most common clinical symptoms include paroxysmal headache, palpitations, and inappropriate sweating. However there are a variety of presenting symptoms that can confuse the diagnosis [1]. Pheochromocytoma can be a life-threatening disease without treatment [2]. Therefore early diagnosis is essential and appropriate treatment must be applied.
- This report presents the case of a patient with pheochromocytoma who had a past medical history of hypertension and diabetes mellitus. We successfully performed surgical resection of a pheochromocytoma associated with a systemic inflammatory syndrome indicated by abnormal liver function tests and severe leukocytosis, and with stress-induced cardiomyopathy presumably due to catecholamine overproduction. Moreover, we successfully treated the patient's uncontrolled diabetes mellitus, and other associated clinical manifestations found at the time of presentation.
INTRODUCTION
- A 49-year-old male presented to our hospital complaining of chest pain. For the past few years the patient reported experiencing intermittent chest pains at night. In addition to the chest pain, the patient developed pyrexia and perspiration beginning a week before admission. He initially visited another hospital for worsening symptoms where examination found abnormal electrocardiography results (the ST elevation in leads aVL and V1 to V6), leukocytosis, and abnormal liver function tests. Echocardiography revealed a decreased cardiac output with regional wall motion abnormality. Abdominal computed tomography (CT) results showed hepatic parenchymal abnormality and a tumor on the left adrenal gland. The patient was transferred to the reporting hospital with these results.
- Seven years prior to presentation the patient had been diagnosed with hypertension and was put on antihypertensive medication, but remained poorly controlled over the 3 years prior to presentation. He was also diagnosed with diabetes mellitus 8 years ago and has been treated with hypoglycemic agents, but his blood sugar was poorly controlled. There was no other medical history.
- On admission, his blood pressure was 140/90 mm Hg with a pulse rate of 66 beats per minute, was breathing at a rate of 20 times per minute, and had a body temperature of 36.8℃. He had clear consciousness without signs of conjunctival pallor, scleral jaundice, dehydration, and absent dilatation of the jugular vein. On chest auscultation breath sounds were clear without wheezing or crackles detected. Cardiac auscultation revealed a heartbeat that was regular without murmur. On examining the abdomen, bowel sounds seemed reduced and the right upper abdomen had slight but not significant tenderness. No masses were detected on abdomen. No limb edema was seen.
- Two days prior to admission, the patient had been tested in another hospital. Blood tests resulted as follows: aspartate aminotransferase (AST) 444 IU/L, alanine aminotransferase (ALT) 469 IU/L, alkaline phosphatase (ALP) 1,153 IU/L, white blood cell count 50,000/µL, hepatitis B surface antigen (HBsAg) negative, anti-HBsAb positive, antihepatitis C virus (anti-HCV) antibody (Ab) negative, antihepatitis A virus (anti-HAV) immunoglobulin M (IgM) Ab negative, and anti-HAV IgG Ab positive. On the day of admission to our hospital, the blood test results were as follows: hemoglobin 15.8 g/dL, white blood cell count 59,360/µL, platelet count 244,000/µL, erythrocyte sedimentation rate 4 mm/hr, and C-reactive protein 3.446 mg/L, blood urea nitrogen 22.7 mg/dL, serum creatinine 0.76 mg/dL, serum calcium 7.2 mg/dL, inorganic phosphorous 2.0 mg/dL, sodium 134 mmol/L, and potassium 4.4 mmol/L. The results also showed that total protein was 5.5 g/dL, albumin was 2.7 g/dL, AST 156 IU/L and ALT 317 IU/L, ALP 289 IU/L, and total bilirubin 1.05 mg/dL. Hemoglobin A1c was 10.7%. At fasting, C-peptide was 0.40 ng/mL (range, 0.48 to 3.3), and 2 hours after a meal it was 0.66 ng/mL. Lactate dehydrogenase was 1,240 IU/L. Creatine kinase MB and troponin-T were found to be 5.99 ng/mL (range, 0.6 to 6.3) and 0.073 ng/mL (range, 0.0 to 0.1), respectively.
- Chest X-ray found no abnormal finding. Abdominal CT at another hospital detected several small-sized and low attenuation lesions on both lobes of the liver, and a 5×5-cm-sized tumor enhanced unevenly on the left adrenal gland (Fig. 1).
- Electrocardiography revealed sinus rhythm with T wave inversion and QT interval prolongation observed in leads II and V1 to V6 (Fig. 2). Although the cardiac ejection fraction was 69%, echocardiography detected severely hypokinetic wall motion of mid-left ventricle sparing basal and apical wall motions.
- The tumor on the left adrenal gland was tested for hormones. The results of 24 hours urinalysis were as follows: vanillylmandelic acid (VMA) 33.93 mg/day (range, 0 to 8.0), metanephrines 20.45 mg/day (range, 0 to 1.3), epinephrine 2,034 µg/day (range, 0 to 40), norepinephrine 978 µg/day (range, 0 to 80), serum epinephrine 1,221 pg/mL (range, 0 to 120), and serum norepinephrine 778 pg/mL (range, 100 to 410). Other tests including aldosterone, plasma renin activity, and cortisol were all normal. On 131I-metaiodobenzylguanidine scan examination, uptake of the radioactive isotope was increased in the left adrenal gland, but no other lesions were found (Fig. 3).
- Based on the examination results above, the patient was diagnosed with a pheochromocytoma on the left adrenal gland. On the 12th day of admission oral phenoxybenzamine administration was started, and blood pressure was followed after having ceased all previous antihypertensives. The dosage of phenoxybenzamine was increased to 35 mg/day. After 19 days of administration, resection of the left adrenal gland was performed under laparoscope. The size of the resected tumor was 5×5 cm, and the surface was a mixture of a hemorrhagic and a cystic lesion. The tissue pathological examination showed the tumor cells with eosinophillic cytoplasm and polygonal shape surrounded by mesenchyma of vasculature on H&E staining, while immunohistochemical stains of chromogranin, synaptophysin, and S-100 immunoperoxidase staining revealed the proper findings to diagnose pheochromocytoma as positive (Fig. 4). Thirteen days after tumor resection a panel of endocrine tests was performed to follow-up on the pheochromocytoma. A 24 hours urinalysis showed a VMA of 6.51 mg/day with metanephrines at 0.61 mg/day, epinephrine at <5 µg/day, norepinephrine at 101.48 µg/day, serum epinephrine at <15 pg/mL, and serum norepinephrine at 536.60 pg/mL (Table 1). When first admitted, the patient had uncontrolled blood pressure even with amlodipine 10 mg and irbesartan 150 mg plus 12.5 mg of hydrochlorothiazide. However, when discharged, the patient had controlled blood pressure with a reduced dosage of antihypertensives consisting of amlodipine 10 mg and irbesartan 150 mg.
- Prior to his admission to the outside hospital, the patient had no history of new drug administration that might cause liver function failure. He also had no history of drinking and had a normal body weight (body mass index 23 kg/m2). A panel for hepatic viral disease (hepatitis B virus, HCV, HAV) was negative, and no abnormality was found through immunological testing (antismooth muscle, antinuclear, antimitochondria antibody), or screening for genetic diseases (plasma iron parameters, ceruloplasmin, Cu). This led to the challenge of finding a clear cause for the liver function failure. The liver function improved with conservative treatment, and had resolved by the time phenoxybenzamine was administered (Table 2). On the 22nd day after left adrenal gland resection, AST and ALT had normalized at 23 and 25 IU/L, respectively (Table 2), and normal liver function test remained within normal limits at 80 days after surgery.
- After admission to our hospital, the patient's leukocytosis continued, and follow-up tests were performed to examine for secondary causes such as blood diseases, infection, and pheochromocytoma. On the fourth day of phenoxybenzamine administration the white blood cell count had recovered to 5,900/µL and continued to be within the normal range even after surgery (Table 2).
- Echocardiography indicated cardiac ejection fraction of 78% on the 15th day after admission (fourth day of phenoxybenzamine administration), and no left ventricle wall motion abnormalities were observed.
- On admission the patient was administered insulin and metformin, but the range of blood glucose fluctuation was wide. However, after surgery blood glucose levels returned to the normal range without the previously administered medications. Previously suppressed C-peptide secretion was recovered 3 days after surgery. On the 80th day after surgery, the hemoglobin A1c was measured found to be 5.7% (Table 1).
CASE REPORT
- Pheochromocytoma can present with various symptoms, and it can be life threatening if diagnosis and treatment are delayed. The patient was suspected to have hyperacute liver failure or systemic inflammatory syndrome on his initial admission, but during the diagnostic process a tumor was found in the left adrenal gland which led to a diagnosis of pheochromocytoma. However, there were confusing features for the case study as follows.
- In this case, the patient had abnormal liver function with clinical symptoms of pheochromocytoma. Norepinephrine overproduction stimulates α adrenalin receptors in the blood vessels which leads to increased resistance of liver arterioles and veins, and decreased blood flow and oxygen volume to the liver [3]. Repeated overproduction can cause damage to hepatocytes which leads to liver function failure [4]. In this case, severely impaired liver function was observed, but looking for the cause did not reveal any abnormal findings. In addition, although the patient's liver function abnormality improved with conservative treatment on admission, and the results returned to the normal range completely after pheochromocytoma surgery (Table 2). Therefore, in this case it is suggested that abnormal liver function could be a secondary change induced by the pheochromocytoma. The previous cases reports mainly showed liver function test results increasing 2- or 3-fold which is not severe. A case Moran et al. [5] reported describes a patient with an AST of 270 IU/L and an ALT of 202 IU/L, but the patient was admitted for chest pain and pyrexia as the chief complaints, diagnosed with myocardial infarction, and only later found to have a pheochromocytoma. This differs from our case since myocardial infarction can be a cause of abnormal liver function.
- In the past, several case reports have described slight and moderate leukocytosis, but only a case from French and Campagna [6] in 1961 and this case report a marked increase of the white cell blood count. French and Campagna [6] confirmed that their patient with perspiration and mandibular pain as chief complaints showed pyrexia (36.6℃ to 39.3℃), and white blood cell count increased to 41,500/mm3. Since there were no abnormalities on examinations causing pyrexia and leukocytosis, it was considered as the secondary changes from pheochromocytoma. Without special treatment the white blood cell count decreased and normalized after surgery. In our case, there were no wide ranges of tumor necrosis that could have caused a marked increase in the white blood cell count. The patient had taken antibiotics for 2 days for pyrexia and pyuria in another hospital he visited, but no signs of pyrexia and pyuria were found in our hospital. Moreover, abdominal CT found multiple lesions on the liver which were confirmed on magnetic resonance imaging, but he was suspected of having a cystic neoplasm or hamartoma since there was insufficient evidence to confirm the condition as infection. In our hospital, urine, sputum, and blood culture tests were performed, all of which were negative. Therefore, leukocytosis was assumed to be a secondary change of pheochromocytoma. If catecholamines are overproduced, lymphocyte and white blood cell count increases through β2-adrenalin receptors [7]. Meanwhile, there are several articles that suggest the leukocytosis could represent a systemic inflammatory response induced by interleukin-6 which is secreted from the pheochromocytoma. Kang et al. [8] published an article in 2005, suggesting that increases in interleukin-6 were accompanied by increased C-reactive protein and fibrinogen, with abnormalities in white blood cell count and liver function tests.
- It is possible that catecholamine overproduction causes various cardiovascular diseases. It is rare to find a pheochromocytoma patient with stress-induced cardiomyopathy, but recent studies have reported these cases. Stress-induced cardiomyopathy is an acute left ventricular dysfunction accompanied by left ventricular regional wall motion abnormality resulting from a sudden emotionally or physically stressful event. Serum epinephrine concentration is raised and functions with the β2-adrenalin receptor to decrease myocardial contractility with a change of signaling from Gs protein to Gi protein in the ventricular cardiac muscle cells. In most cases, left ventricle dysfunction is reversible and has positive consequences [9]. In this case study, on the day of admission echocardiography showed the patient's cardiac ejection fraction was decreased and left ventricular regional wall motion abnormality was observed. However, abnormal cardiac function improved with improvement of other pheochromocytoma-related parameters. The patient's electrocardiography found ST segment elevation at the outside hospital 3 days prior to the admission; however, the pattern was not typical of acute coronary syndrome. Troponin-T is a well-known marker which usually persists elevated level for 7 days after myocardiac infarction. However, it was within the normal range when the patient was admitted to our hospital. For these reasons, he was diagnosed with stress-induced cardiomyopathy rather than acute coronary syndrome.
- A Japanese case study reported that 38% of pheochromocytoma patients have diabetes and 51% have impaired glucose tolerance [10]. When epinephrine is secreted, production of glucose is increased and consumption is decreased in the liver under a β adrenalin mediated mechanism [11]. Meanwhile, catecholamines affect α2-adrenalin receptors to lower the secretion of insulin [12].
- There are several cases suggesting that after pheochromocytoma has been treated by surgical resection, symptoms of diabetes and impaired glucose tolerance improve [13]. However, there are rare cases reporting a full recovery of diabetes. The domestic cases reporting full recovery of diabetes mellitus with treatment of pheochromocytoma only includes a case reported by Hong et al. [14] in 2001 and a case by Cho et al. [15] in 2007. Hong et al. [14] reported that a diabetic patient medicated with oral hypoglycemic agents for 2 years (Hemoglobin A1c, 7.5%) was completely cured after pheochromocytoma treatment. Cho et al. [15] reported on a diabetic patient diagnosed 6 months previously for hyperglycemia (hemoglobin A1c, 15%) and then coincidently diagnosed with pheochromocytoma. The patient was completely cured after treating the pheochromocytoma. In comparison, the patient in this case had diabetes mellitus for 8 years, and insulin and oral hypoglycemic agents were administered due to elevated blood glucose just before admission. However, it is significant that diabetes in this patient showed complete remission after pheochromocytoma surgery. The patient had a hemoglobin A1c of 10.7% before surgery, but after surgery and without the use of oral hypoglycemic agents and insulin, his blood glucose, hemoglobin A1c, and C-peptide normalized.
- In this case, a follow-up test on the 13th day after surgery showed a decreased, but still slightly higher than normal level of norepinephrine in the serum and secreted in the urine. However, according to previous reports, the diagnostic specificity of norepinephrine for pheochromocytoma varies between 81% to 100% [16,17,18,19]. Additionally, a study of receiver operating characteristic curve analysis on diagnosis showed that the 24 hours urinalysis of total metanephrines is more useful for diagnosing pheochromocytoma than the 24 hours urine catecholamines [19], and some studies reported that serum catecholamines are less useful than other examinations such as 24 hours urinalysis of VMA, catecholamines, or free metanephrines [20]. In this case report, the clinical condition and accompanying secondary diseases of this patient improved after surgery. The fact that total urinary metanephrines, a more sensitive test for diagnosing pheochromocytoma, became normal suggests that no remaining tumor exists. However, follow-up studies are necessary to monitor the possible recurrence of pheochromocytoma.
- Thus, pheochromocytoma can be cured completely with accurate medicinal and surgical treatment after diagnosis, and its related clinical conditions tend to be reversible. When treating patients with typical symptoms of pheochromocytoma accompanied by various clinical symptoms related to catecholamine overproduction, it is important to include pheochromocytoma on the differential diagnosis and diagnose at an early stage.
- Pheochromocytoma can present with various clinical symptoms and can be life-threatening if not treated. However, early diagnosis and accurate treatment can bring a high rate of complete cure. Moreover, accompanying clinical conditions tend to be reversible. The authors report a case of a patient with pheochromocytoma accompanied by rare clinical symptoms such as a systemic inflammatory syndrome with abnormal liver function tests and severe leukocytosis, and stress-induced cardiomyopathy induced by catecholamine overproduction. The patient also had a history of hypertension and diabetes mellitus that resolved with surgical resection of the pheochromocytoma.
DISCUSSION
- 1. Lenders JW, Eisenhofer G, Mannelli M, Pacak K. Phaeochromocytoma. Lancet 2005;366:665–675. ArticlePubMed
- 2. Brouwers FM, Lenders JW, Eisenhofer G, Pacak K. Pheochromocytoma as an endocrine emergency. Rev Endocr Metab Disord 2003;4:121–128. ArticlePubMed
- 3. Greenway CV, Lautt WW. Distensibility of hepatic venous resistance sites and consequences on portal pressure. Am J Physiol 1988;254(3 Pt 2):H452–H458. ArticlePubMed
- 4. Wang P, Tait SM, Chaudry IH. Sustained elevation of norepinephrine depresses hepatocellular function. Biochim Biophys Acta 2000;1535:36–44. ArticlePubMed
- 5. Moran ME, Rosenberg DJ, Zornow DH. Pheochromocytoma multisystem crisis. Urology 2006;67:846Article
- 6. French C, Campagna FA. Pheochromocytoma with shock, marked leukocytosis, and unusual electrocardiograms. Case report and review of the literature. Ann Intern Med 1961;55:127–134. ArticlePubMed
- 7. Benschop RJ, Rodriguez-Feuerhahn M, Schedlowski M. Catecholamine-induced leukocytosis: early observations, current research, and future directions. Brain Behav Immun 1996;10:77–91. ArticlePubMed
- 8. Kang JM, Lee WJ, Kim WB, Kim TY, Koh JM, Hong SJ, Huh J, Ro JY, Chi HS, Kim MS. Systemic inflammatory syndrome and hepatic inflammatory cell infiltration caused by an interleukin-6 producing pheochromocytoma. Endocr J 2005;52:193–198. ArticlePubMed
- 9. Lyon AR, Rees PS, Prasad S, Poole-Wilson PA, Harding SE. Stress (Takotsubo) cardiomyopathy: a novel pathophysiological hypothesis to explain catecholamine-induced acute myocardial stunning. Nat Clin Pract Cardiovasc Med 2008;5:22–29. ArticlePubMedPDF
- 10. Seino Y, Imura H. Endocrine disorders and diabetes in Japan. Diabetes Res Clin Pract 1994;24(Suppl):S129–S133. ArticlePubMed
- 11. Rizza RA, Cryer PE, Haymond MW, Gerich JE. Adrenergic mechanisms for the effects of epinephrine on glucose production and clearance in man. J Clin Invest 1980;65:682–689. ArticlePubMedPMC
- 12. Isles CG, Johnson JK. Phaeochromocytoma and diabetes mellitus: further evidence that alpha 2 receptors inhibit insulin release in man. Clin Endocrinol (Oxf) 1983;18:37–41. ArticlePubMed
- 13. Wiesner TD, Bluher M, Windgassen M, Paschke R. Improvement of insulin sensitivity after adrenalectomy in patients with pheochromocytoma. J Clin Endocrinol Metab 2003;88:3632–3636. ArticlePubMed
- 14. Hong CK, Ahn YB, Kim SH, Woo YS, Lee SG, Ko SH, Song HK, Yoon KH, Kang MI, Cha BY, Lee KW, Son HY, Kang SK. A case of cured diabetes mellitus after pheochromocytoma removal. J Korean Soc Endocrinol 2001;16:502–507.
- 15. Cho HC, Kim HS, Kim YJ, Hah YJ, Kim NK, Kim MK, Park KG, Kim YH, Kwon SY. A case of cured diabetes mellitus after occult malignant pheochromocytoma removal. J Korean Diabetes Assoc 2007;31:520–524.Article
- 16. Lenders JW, Pacak K, Walther MM, Linehan WM, Mannelli M, Friberg P, Keiser HR, Goldstein DS, Eisenhofer G. Biochemical diagnosis of pheochromocytoma: which test is best? JAMA 2002;287:1427–1434. ArticlePubMed
- 17. Tormey WP, FitzGerald RJ. Lack of uniformity in the clinical approach to the interpretation of urinary catecholamines and their metabolites. Ir J Med Sci 1995;164:146–150. ArticlePubMedPDF
- 18. Raber W, Raffesberg W, Bischof M, Scheuba C, Niederle B, Gasic S, Waldhausl W, Roden M. Diagnostic efficacy of unconjugated plasma metanephrines for the detection of pheochromocytoma. Arch Intern Med 2000;160:2957–2963. ArticlePubMed
- 19. Kudva YC, Sawka AM, Young WF Jr. Clinical review 164: the laboratory diagnosis of adrenal pheochromocytoma: the Mayo Clinic experience. J Clin Endocrinol Metab 2003;88:4533–4539. ArticlePubMedPDF
- 20. Boyle JG, Davidson DF, Perry CG, Connell JM. Comparison of diagnostic accuracy of urinary free metanephrines, vanillyl mandelic acid, and catecholamines and plasma catecholamines for diagnosis of pheochromocytoma. J Clin Endocrinol Metab 2007;92:4602–4608. ArticlePubMedPDF
References
Fig. 1Abdominal computed tomography scan shows the 5×5-cm-sized, heterogeneous mass in left adrenal gland (white arrow).
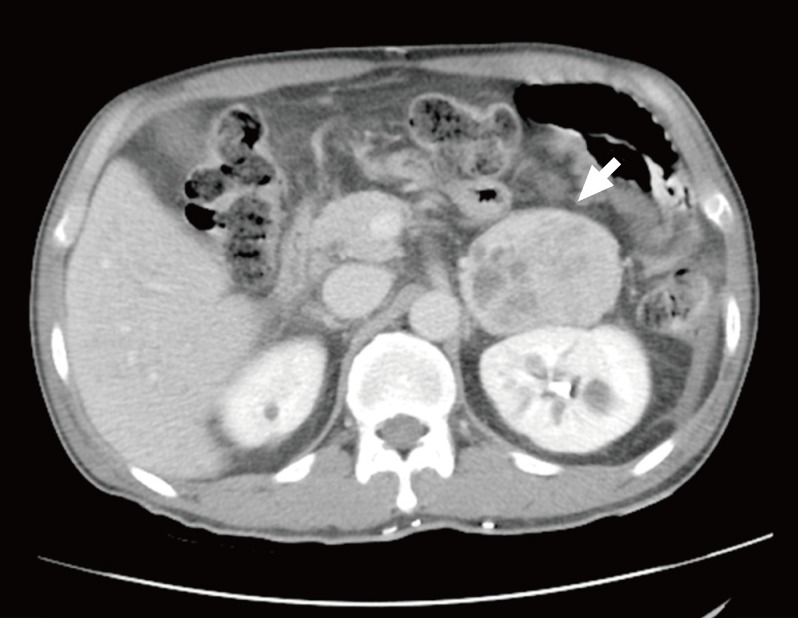

Fig. 3
131I-Metaiodobenzylguanidine (MIBG) scan shows an increased uptake in the left upper abdomen.

Fig. 4Histopathology of pheochromocytoma. (A) The tumor cells with eosinophillic cytoplasm and polygonal shape, are surrounded by mesenchyma of vasculature (H&E stain, ×200). Cromogranin (B, ×200), synaptophysin (C, ×200), and S-100 (D, ×200) are stained.
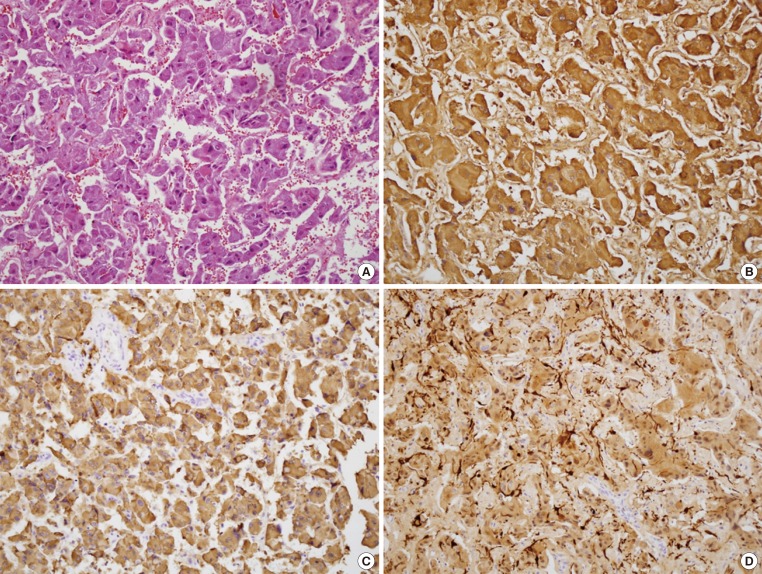
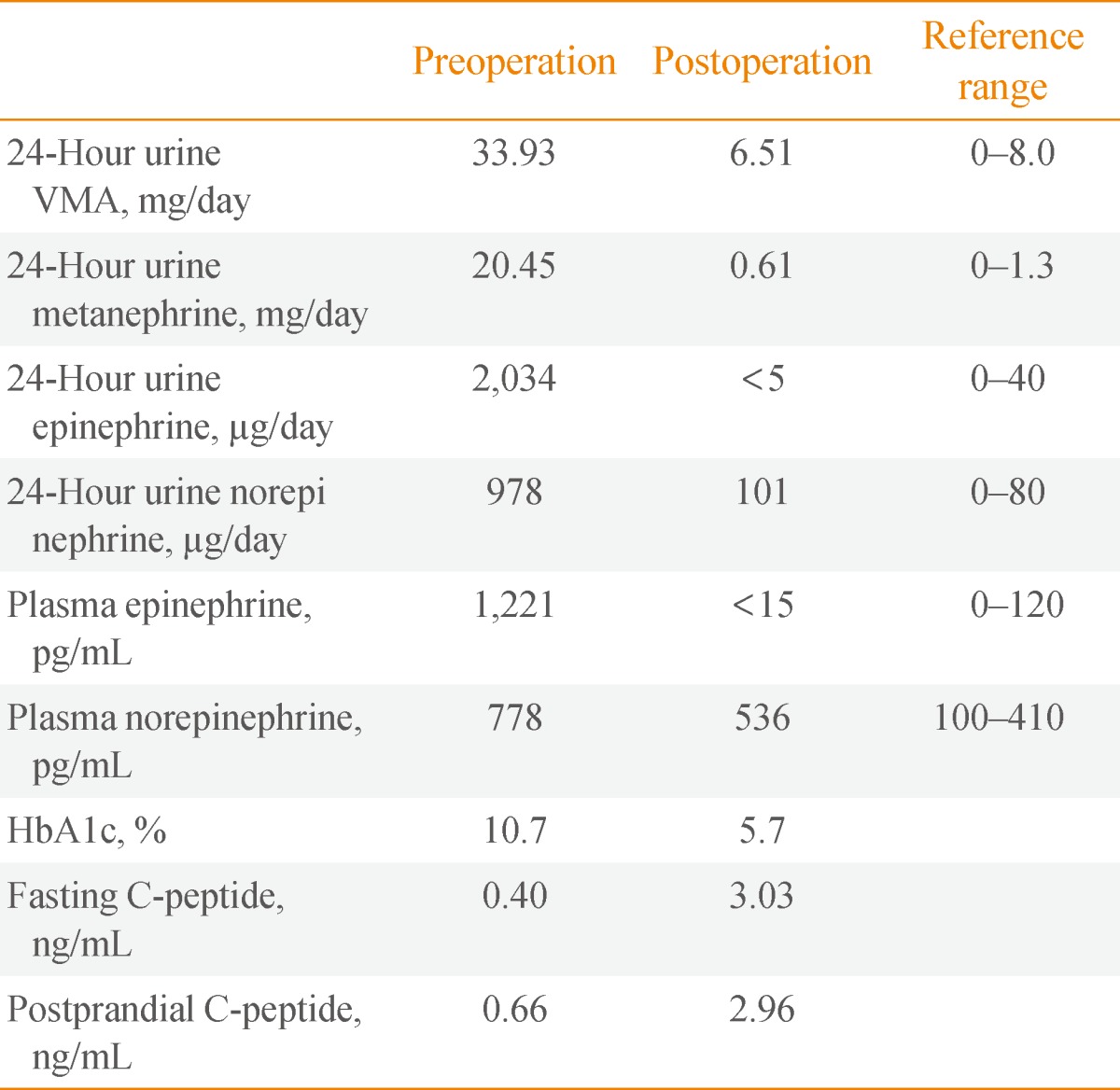
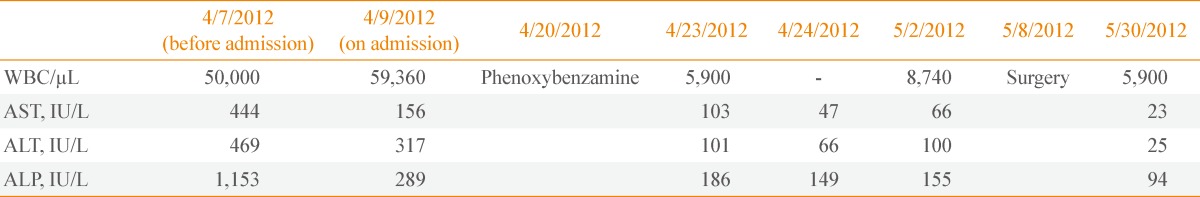
Figure & Data
References
Citations
Citations to this article as recorded by 
- Liver Transplantation for Fulminant Hepatic Failure Precipitated by Pheochromocytoma Crisis in the Setting of Using Garcinia cambogia Weight Loss Supplement: A Case Report
Motaz A Selim, Krystal Weierstahl, Calvin Eriksen, Terra Pearson, Harvey Woehlck
Cureus.2023;[Epub] CrossRef - Pheochromocytoma presenting with severe abdominal pain and abnormal liver enzymes
Mhd Baraa Habib, Mohamed Abdelrazek, Sali Alatasi, Mouhand F. H. Mohamed, Hamda Ali, Mohamad Khair Hamad
Clinical Case Reports.2021;[Epub] CrossRef - A cardiogenic shock in PICU turned out to be a bilateral pheochromocytoma: A case report
Jay Shah, Zinkal Patel, Shradha Patel, Amit Kumar
Journal of Pediatric Critical Care.2021; 8(5): 255. CrossRef - Endocrine Diseases and the Liver
Miguel Malespin, Ammar Nassri
Clinics in Liver Disease.2019; 23(2): 233. CrossRef - Pheochromocytoma crisis presenting with hypotension, hemoptysis, and abnormal liver function
Renhua Wu, Nanwei Tong, Xinlei Chen, Shishi Xu, Fang Zhang, Lizhi Tang, Yuwei Zhang
Medicine.2018; 97(25): e11054. CrossRef - Articles in 'Endocrinology and Metabolism' in 2014
Won-Young Lee
Endocrinology and Metabolism.2015; 30(1): 47. CrossRef
 PubReader
PubReader Cite
Cite




