Articles
- Page Path
- HOME > Endocrinol Metab > Volume 37(1); 2022 > Article
-
Review ArticleThyroid Development of Metabolic Synthetic Lethality and Its Implications for Thyroid Cancer
-
Sang-Hyeon Ju1
 , Seong Eun Lee2, Yea Eun Kang1, Minho Shong1
, Seong Eun Lee2, Yea Eun Kang1, Minho Shong1 -
Endocrinology and Metabolism 2022;37(1):53-61.
DOI: https://doi.org/10.3803/EnM.2022.1402
Published online: February 28, 2022
1Division of Endocrinology and Metabolism, Department of Internal Medicine, Chungnam National University College of Medicine, Daejeon, Korea
2Department of Medical Science, Chungnam National University College of Medicine, Daejeon, Korea
- Corresponding author: Minho Shong. Division of Endocrinology and Metabolism, Department of Internal Medicine, Chungnam National University College of Medicine, 282 Munhwa-ro, Jung-gu, Daejeon 35015, Korea, Tel: +82-42-280-7161, Fax: +82-42-280-6990, E-mail: minhos@cnu.ac.kr
• Received: January 13, 2022 • Revised: January 26, 2022 • Accepted: January 27, 2022
Copyright © 2022 Korean Endocrine Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (https://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
- Cancer therapies targeting genetic alterations are a topic of great interest in the field of thyroid cancer, which frequently harbors mutations in the RAS, RAF, and RET genes. Unfortunately, U.S. Food and Drug Administration-approved BRAF inhibitors have relatively low therapeutic efficacy against BRAF-mutant thyroid cancer; in addition, the cancer often acquires drug resistance, which prevents effective treatment. Recent advances in genomics and transcriptomics are leading to a more complete picture of the range of mutations, both driver and messenger, present in thyroid cancer. Furthermore, our understanding of cancer suggests that oncogenic mutations drive tumorigenesis and induce rewiring of cancer cell metabolism, which promotes survival of mutated cells. Synthetic lethality (SL) is a method of neutralizing mutated genes that were previously considered untargetable by traditional genotype-targeted treatments. Because these metabolic events are specific to cancer cells, we have the opportunity to develop new therapies that target tumor cells specifically without affecting healthy tissue. Here, we describe developments in metabolism-based cancer therapy, focusing on the concept of metabolic SL in thyroid cancer. Finally, we discuss the essential implications of metabolic reprogramming and its role in the future direction of SL for thyroid cancer.
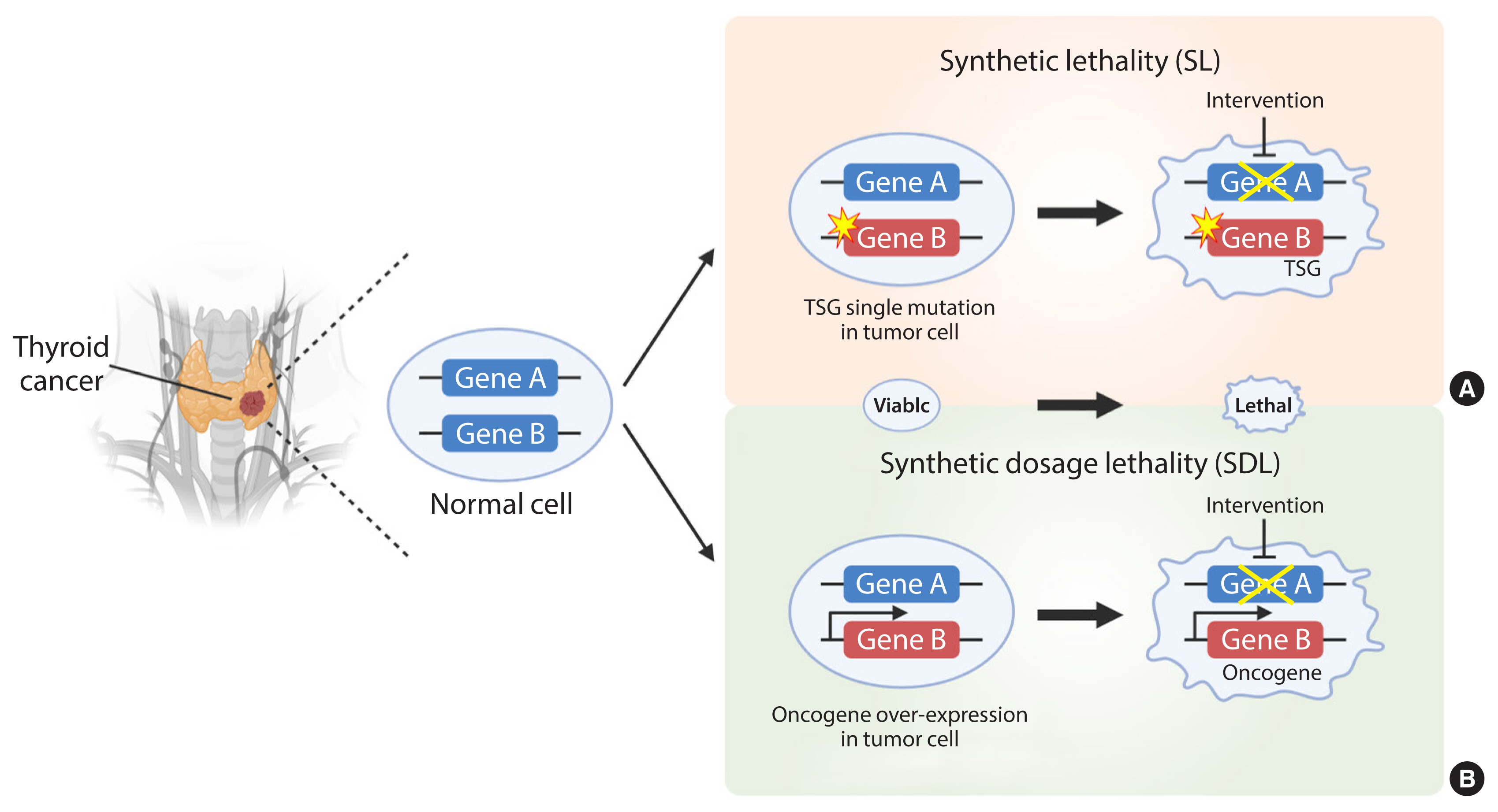
- Synthetic lethality (SL), first proposed in 1922 by the American geneticist Calvin Bridges, is observed when simultaneous inhibition of two genes results in cellular lethality [1]. The meaning of synthetic used here conforms to the ancient Greek meaning: “the combination of entities to form something new.” Classical SL occurs when inhibition of two genes is lethal, while inhibition of either gene alone is not (Fig. 1). It can be harnessed to treat cancer selectively by identifying inactive genes and targeting their SL partner(s) [2]. In accordance with this concept, SL can also be applied to genes and small molecules used to elucidate the mode of action of drugs [3–5]. Cancer is driven by consequences of mutation or amplification of genes that allow cells to escape mechanisms that control proliferation and survival. Hence, inhibitors that target SL partners of mutated or amplified genes in cancer cells can cause cancer cell death without affecting the survival of normal cells.
- The SL concept is divided into two categories due to its complexity: SL and synthetic dosage lethality (SDL); the latter is a slight deviation from the original concept of SL (Fig. 1). SL occurs in cancers caused by loss-of-function of tumor suppressors and their partner genes. This is a genetic interaction in which combination of two mutations leads to cell death, whereas a single mutation in either of the genes does not. SDL occurs during interaction between an oncogene and its interactor gene [6]. Principally, SL and SDL offer a unique opportunity to develop selective anticancer drugs [7,8].
- Recent studies identified SL interactions via systematic screening of human cancer cells using small molecule inhibitors and high-throughput short hairpin RNA (shRNA) or Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/Cas9-based screens [9,10]. These screens have been used successfully to identify SL pathways associated with known tumor suppressors and oncogenes [11]. Therapeutics developed using the SL approach have been used to treat breast cancer. Loss-of-function of breast cancer susceptibility genes 1 and 2 (BRCA1 and BRCA2), coupled with inhibition of poly (adenosine diphosphate [ADP]-ribose) polymerase (PARP), is an SL interaction that results in marked sensitization of cancer cells [12,13]. Fusion oncogenes such as RET/papillary thyroid cancer (PTC) and breakpoint cluster region-Abelson (BCR-ABL) drive carcinogenesis in thyroid cancer and chronic myelogenous leukemia (CML). Addiction of tumor cells to constitutively active RET/PTC and BCR-ABL protein kinases results in sensitization to kinase inhibitors such as imatinib [14–16]. Recently, screening identified an SL interaction between signal transducer and activator of transcription 3 (STAT3) and BCR-ABL, such that combined inhibition of STAT3 and BCR-ABL1 induced SL in therapy-resistant CML [17].
- Furthermore, targeting undruggable oncogenes such as KRAS mutations, which are common in refractory thyroid cancer, can be approached via SL interactions [18–20]. MYC oncogenes are implicated in the development of a large number of cancers, including thyroid cancer [21]. Although MYC is an attractive target for cancer therapeutics, lack of defined pockets in the MYC protein mean that it is regarded traditionally as undruggable by low molecular weight inhibitors [22]. A recent study aimed at identifying SL partners of oncogenic MYC revealed that the most over-represented functional categories among MYC SL genes are DNA-repair and the cell cycle [23–26]. Elucidation of KRAS and MYC synthetic lethal interactions is still in its infancy, and how these interactions are influenced by tissue-specific programs and by concurrent genetic changes requires further investigation. Nevertheless, we predict that these studies may lead to novel therapeutic approaches and new insights into the role of KRAS and MYC in thyroid cancers that are refractory on radioiodine and kinase inhibitors.
INTRODUCTION
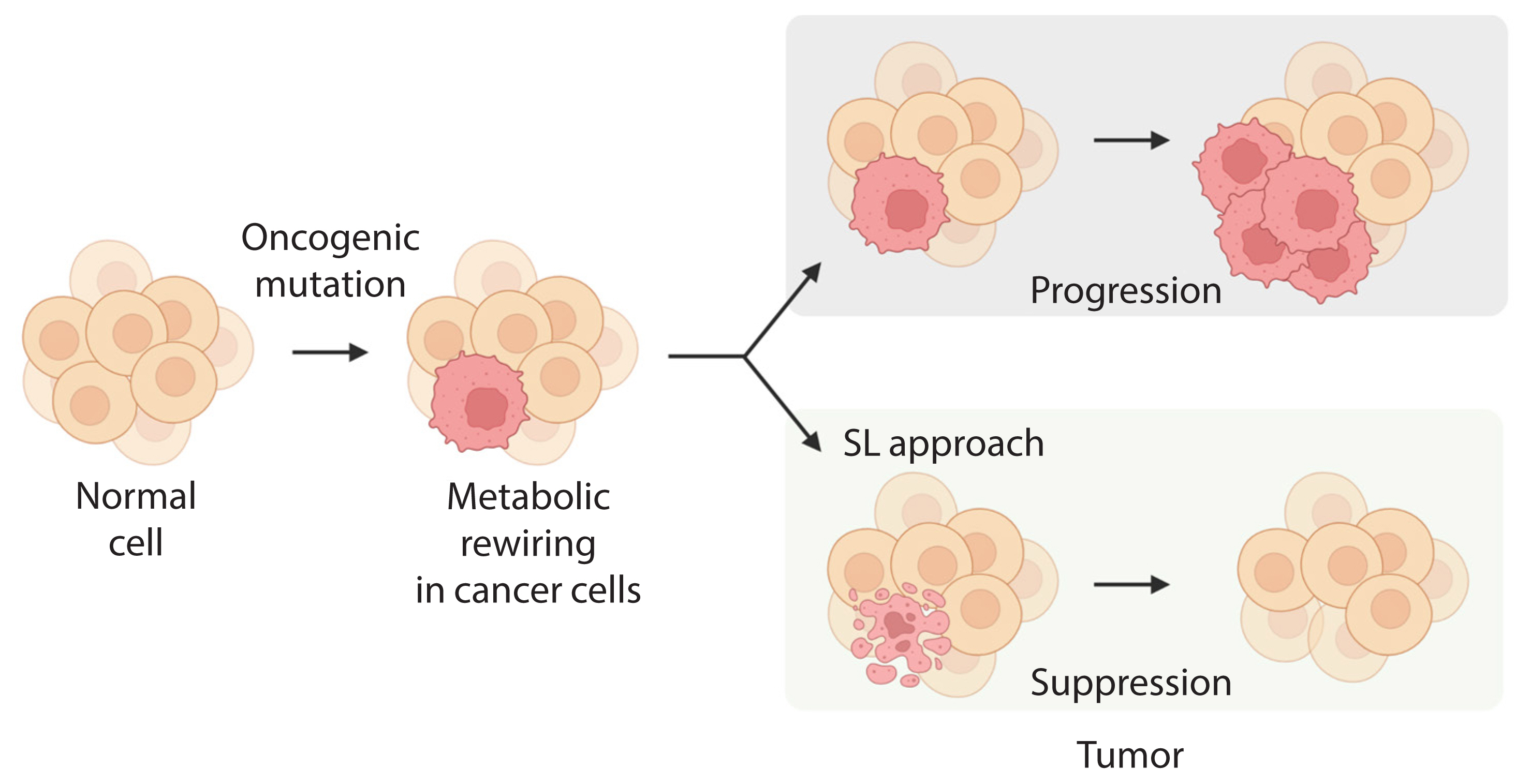
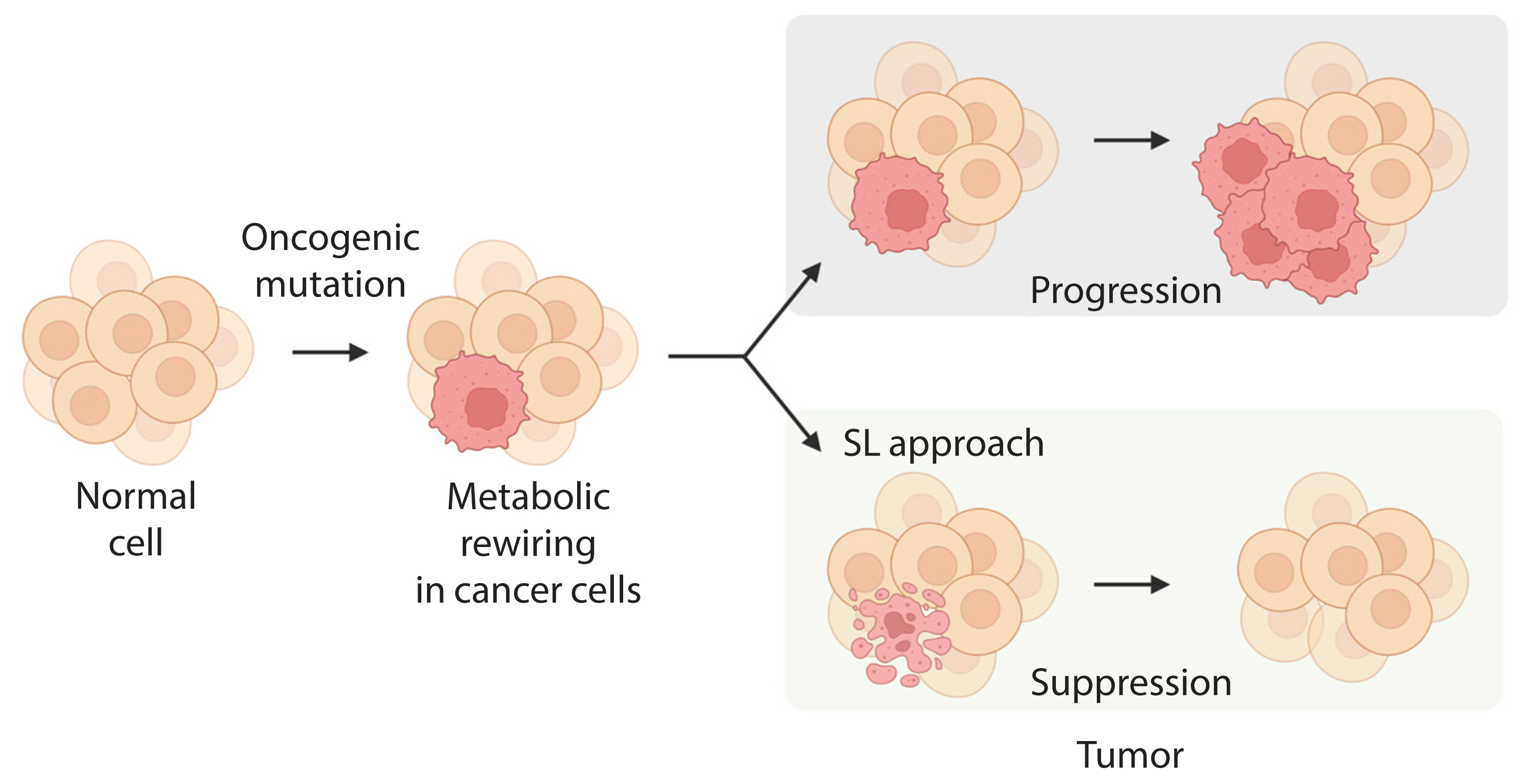
- Altered cellular metabolism is a hallmark of cancer, and a deeper understanding of cancer cell metabolism may provide the basis for metabolism-based cancer therapy. This concept was encouraged by the finding that oncogene mutations, or mutations in key metabolic enzymes, drive tumorigenesis by reprogramming or rewiring cell metabolism required for dysregulated cell growth and proliferation [27]. Cancer cells largely use three key metabolic processes, glycolysis, amino acid catabolism, and mitochondrial oxidative phosphorylation (OXPHOS), to adapt to the increased energy demand required to drive growth and metastasis [27,28]. Recently, the concept of SL was expanded to include cellular metabolism, and it was applied to cancer-specific metabolic remodeling (Fig. 2) [29–32]. For example, AMPK-related kinase 5 (ARK5), an upstream regulator of AMP-activated protein kinase (AMPK), is an SL interactor with oncogenic MYC via activation of the mammalian target of rapamycin (mTOR) pathway [25]. In oncogenic MYC-expressing cells, inhibition of ARK5 leads to a collapse in adenosine triphosphate (ATP) levels and results in induction of pro-apoptotic responses. In addition, depletion of ARK5 in MYC-driven mouse models of hepatocellular carcinoma prolongs survival and demonstrates the therapeutic value of this SL interaction.
- Enhanced glucose uptake and an increased rate of glycolysis are major remodeled metabolic features of cancer cells, including differentiated and undifferentiated thyroid cancers. Increased flux of glucose into cancer cells generates high levels of lactate from pyruvate through lactate dehydrogenase A (LDHA) [33,34]. Blockade of LDHA is not sufficient to inhibit cancer cells, but it remodels amino acid biosynthesis by engaging activating transcription factor 4 (ATF4)-dependent metabolic reprogramming and by inducing solute carrier family 1 member 5 (SLC1A5) to increase uptake of glutamine and essential amino acids, thereby enhancing mammalian target of rapamycin complex 1 (mTORC1) activity and cell proliferation. Co-targeting LDHA and mTORC1 has a synergistic effect with respect to suppressing melanoma cell proliferation and tumor growth [35]. In addition, activation of the LDHA pathway reduces nicotinamide adenine dinucleotide (NAD+) to NADH, which is used to transport electrons to the electron transport chain (ETC). The combination of LDHA inhibition with small inhibitors FX11 and FK866 (NAD+ synthesis inhibitors) reduces the NAD+ cellular pool in lymphoma, resulting in tumor regression [36]. Dual inhibition of glycolysis and mitochondrial function using a combination of 2-deoxyglucose, a specific glycolysis inhibitor, and metformin impairs tumor growth in mouse xenograft models [37,38]. Recently, Benjamin et al. [39,40] showed that syrosingopine, a dual monocarboxylate transporter 1 (MCT1) and MCT4 inhibitor that prevents lactate and H+ efflux, elicits SL in combination with metformin. This finding indicates that loss of NAD+ regeneration capacity due to combined treatment with metformin and syrosingopine results in glycolytic blockade, leading to ATP depletion and cell death. A previous study shows that metformin has an in vitro synergistic anticancer effect against anaplastic thyroid cancer when used in combination with vemurafenib [41].
- The serine synthesis pathway and one-carbon metabolism are often dysregulated in cancer, including thyroid cancer, due to their contribution to cellular building blocks, DNA methylation, post-translational modification, and redox homeostasis [42]. The complex networks of enzymes such as phosphoglycerate dehydrogenase (PHGDH), serine hydromethyl transferase (SHMT), and methylenetetrahydrofolate dehydrogenase (MTHFD), which are required for the serine synthesis pathway and one-carbon metabolism in the cytoplasm and mitochondria, may provide a target for SL in thyroid cancer [43].
- SL screens targeting metabolic enzymes could be useful tools that might add to the number of metabolic targets for anticancer therapy, and they could be exploited to overcome resistance to conventional chemotherapy agents such as bevacizumab (Avastin, Genentech Inc., San Francisco, CA, USA). A potential target is glycogen phosphorylase liver form (PYGL), expression of which increased following bevacizumab treatment in an in vivo xenograft model [44]. Combination treatment with inhibitors targeting PYGL and/or other hypoxia-inducible factor (HIF)-dependent metabolic enzymes essential for survival under hypoxic conditions, plus an anti-angiogenic drug, could be of great interest as a metabolic cancer therapy.
- Alteration of one single pathway in the metabolic network may induce compensatory pathways to generate alternative sources that compensate for the loss of certain metabolites. Metabolic SL occurs when simultaneous mutations in two different metabolic genes are lethal, while a mutation in either of the individual genes alone is tolerable. The simultaneous suppression of several genes that together have the potential for SL may open new avenues for anticancer treatment. The genome-scale network model of cancer metabolism, shRNA, and CRISPR/Cas9 screening to validate SL, will be used widely to identify genes essential for cancer cell proliferation. Furthermore, they will predict combinations of SL drug targets that can be used to treat thyroid cancer.
- Understanding the context-dependent functions of SL allows therapeutic targeting of cancer. Furthermore, elucidating SL gene combinations could identify clinically relevant drug combinations, as well as biomarkers, that will allow development of improved treatments. The recent SL gene combinations used in preclinical and clinical settings may facilitate development of new therapeutic approaches and identify potential biomarkers [45].
METABOLIC SYNTHETIC LETHALITY
- The BRAFV600E mutation is the most common genetic alteration in thyroid cancer, occurring in about 45% of sporadic PTCs, particularly relatively aggressive subtypes such as the tall-cell PTC [46]. The BRAFV600E mutant protein has 500-fold greater kinase activity than the wild-type [47], and this mutation is mutually exclusive with other common genetic alterations, supporting its independent oncogenic role (as demonstrated by targeted expression studies showing BRAF mutation-initiated development of PTC and its transition to anaplastic thyroid cancer) [48,49].
- Several tyrosine kinase inhibitors have been approved for treatment of differentiated and anaplastic thyroid cancers. Lenvatinib and sorafenib are used widely to treat radioiodine-refractory differentiated thyroid cancer. However, although these targeted therapies extend median progression-free survival, they do not increase overall survival [50]. A significant number of severe side effects, as well as development of drug resistance, prevent effective and long-term treatment. Additional studies are needed to characterize the molecular and metabolic landscape, and to identify a potential effective therapy for aggressive BRAFV600E mutant thyroid cancers.
- The high prevalence of the BRAF mutation makes it an attractive SDL lethal target due to possible signaling dependencies consequent to hyperactivation of the mitogen-activated protein kinase (MAPK) pathway. Transcription factor networks downstream of MAPK, as well as cataloged MAPK target genes encoding nuclear DNA-binding proteins, have been identified in colorectal cancer cell lines [51,52]. Klotz-Noack et al. [53] performed a shRNA screen to probe MAPK targets encoding nuclear and/or DNA-binding factors in isogenic colorectal cell lines inducible for oncogenic BRAF. They identified the multifunctional Splicing Factor Proline and Glutamine Rich Protein (SFPQ) as a novel factor that is SL with BRAFV600E. Depletion of SFPQ from BRAFV600E-expressing, but not isogenic BRAF-wild-type, colorectal cancer cells halted cell cycle progression at S phase and triggered apoptotic cell death [53]. Mechanistically, loss of SFPQ from BRAF-mutant cancer cells triggers the Chk1-dependent replication checkpoint, resulting in decreased cell numbers and reduced activity of replication factories, thereby increasing collision between replication and transcription [53]. BRAFV600E-mutant cancer cells and organoids are sensitive to combinations of Chk1 inhibitors and chemically-induced replication stress, pointing toward future therapeutic approaches that exploit nuclear vulnerabilities induced by BRAFV600E [53]. This screening approach highlights an underused approach to targeted therapeutic interference in BRAF-mutant colorectal cancer cells by exploiting the essential nuclear programs induced by MAPK activity identified in thyroid cancer cell lines. Consistent with the results in colon cancer cell lines, low expression of SFPQ is associated with decreased survival probability in The Cancer Genome Atlas (TCGA) dataset [54]. Further investigation of the role of SFPQ in the context of BRAFV600E-mutant thyroid cancer cells is required. Publicly available cancer cell lines and tumor tissue genomic data used to look for SDL partners of mutant BRAF identified limited SL pairs [55]. The genes PCDHGB4, ZNF138, CXCR2, CDH2, and DGKA were suggested as potential SL genes in BRAF-mutant cancer cells [55].
- Li et al. [56] identified cytochrome P450 family 2 subfamily S member 1 (CYP2S1) as a SL partner of BRAFV600E in thyroid cancer. They found that expression of CYP2S1 in PTCs, particularly in conventional and tall-cell PTCs, is higher than that in normal thyroid tissues, and that its expression is positively associated with the BRAFV600E mutation. Consistent with this, knockdown of CYP2S1 selectively inhibits cell proliferation, migration, invasion, and tumorigenic potential in nude mice, and promotes cell apoptosis in BRAFV600E-mutated thyroid cancer cells, but not in BRAF wild-type lines. Mechanistically, the BRAFV600E-mediated MAPK/extracellular signal-regulated kinase (ERK) cascade upregulates CYP2S1 expression via an aryl hydrocarbon receptor (AHR)-dependent pathway, while CYP2S1 in turn enhances the transcriptional activity of AHR through its metabolites [56].
- The metabolic remodeling induced by BRAFV600E expression can also be exploited as a SL vulnerability [57]. Use of an shRNA screen selectively targeting genes involved in ketogenic metabolism in BRAFV600E and BRAF wild-type melanoma cell lines identified 3-hydroxy-3-methylglutaryl-CoA lyase (HMGCL) as a possible SL partner for BRAFV600E [57]. Increased levels of HMGCL, an enzyme involved in ketogenesis, were observed in BRAFV600E melanomas in which HMGCL was knocked down, resulting in decreased growth solely in BRAFV600E mutant lines. Mechanistically, acetoacetate (the metabolite created by HMGCL) increases binding of BRAFV600E to MAPK kinase (MEK), with subsequent phosphorylation and activation of the MAPK pathway [57]. Suppression of HMGCL specifically attenuates proliferation and growth potential caused by decreased MAPK pathway activity. Data from the human protein atlas dataset show that thyroid cancer has weak to moderate cytoplasmic immunoreactivity for HMGCL. Therefore, the susceptibility of BRAFV600E thyroid cancer cells to inhibition of HMGCL remains to be determined.
- BRAF-induced senescence is a tumor suppressive mechanism that prevents transformation in response to constitutive oncogene activation. Activation of oncogenes also promotes metabolic reprogramming to support cancer cell metabolism, but the role of metabolic rewiring in BRAFV600E-induced senescence remains to be identified. Kaplon et al. [58] showed that cells undergoing BRAFV600E-induced senescence exhibited increased incorporation of pyruvate into the tricarboxylic acid (TCA) cycle, as well as increased redox stress. This increase in mitochondrial oxidative metabolism in response to BRAFV600E-induced senescence was mediated by activation of pyruvate dehydrogenase (PDH), a gatekeeper enzyme that links glycolysis to the TCA cycle and is regulated by phosphorylation by pyruvate dehydrogenase kinase (PDK). Induction of PDH activity in BRAFV600E-induced senescent cells is associated with downregulation of PDK1. This, and the results of another study, suggest that PDK1 inhibitors may synergize with BRAF inhibitors to trigger melanoma cell death [59].
- Porchia et al. [60] demonstrated that the compound OSU-03012, a novel PDK1 inhibitor, inhibits thyroid cancer cell proliferation and migration through multiple pathways involved in thyroid cancer progression. However, this compound needs to be validated as an inducer of SL in BRAFV600E-induced thyroid cancer.
BRAF MUTATION AND SDL
- The genetic basis of cancer progression has been studied extensively. By contrast, the role of mitochondrial genomic mutations in development of thyroid cancer has not. The majority of mitochondrial proteins are encoded by nuclear DNA, with only 37 genes encoded by mitochondrial DNA (mtDNA). These include 13 components of the ETC, 22 transfer RNAs, and two ribosomal RNAs. Several studies show that acquired mtDNA alterations are associated with thyroid cancer. It has been known for over three decades that thyroid tumors contain abnormally high numbers of mitochondria. The majority of the mutations were found in mitochondrial complex I of the respiratory chain, and severe functional defects in complex I activity were observed in thyroid tumor cell lines [61,62].
- In addition to the clear genetic alterations in TCA cycle enzymes, various types of cancer display heterogenous mitochondrial OXPHOS function. Therefore, SL strategies based on metabolic reprogramming triggered by these mitochondrial OXPHOS-proficient and -deficient cancers have been proposed. For instance, cancer harboring mutations in TCA cycle enzymes fumarate hydratase (FH) or succinate dehydrogenase (SDH) represent OXPHOS-deficient cancer subtype [63]. Recently, Sun et al. [64] identified metabolic genes that are synthetically lethal with OXPHOS deficiency caused by a defect in FH. Genetic loss-of-function screening found that inhibition of phosphogluconate dehydrogenase (PGD) blocks proliferation of FH mutant cancer cells. It was found that FH mutant cancer cells that exhibit defects in the respiratory chain are sensitive to glycolysis and reductive glutamine carboxylation, which normally lead to activation of respiration [64].
- As well as identifying multiple cancers that are highly OXPHOS-proficient, Molina et al. [31] discovered an inhibitor, IACS-010759, which is specifically effective in cancer cells that are deficient in glycolysis. IACS-010759 is an inhibitor of complex I of the mitochondrial ETC. IACS-010759 inhibited the oxygen consumption rate and cell growth by up to 100% in several cancer (pancreatic, lung, ovarian, and breast) cell lines. Glycolysis and OXPHOS co-operate to maintain the cellular energetic balance, and so genetic or pharmacological inhibition of OXPHOS can result in compensatory upregulation of glycolysis to maintain ATP levels. They hypothesized that tumor cells with a reduced capacity for glycolysis would thus be more sensitive to OXPHOS inhibition.
- Thyroid cancer cells also showed heterogeneity with respect to OXPHOS function, which can be regulated by genomic alterations, mtDNA mutations, fusion and fission abnormalities, mitophagy, and mitochondrial biogenesis. Application of SL based on OXPHOS function in thyroid cancer requires profiling of mitochondrial function in patients with thyroid cancer.
MITOCHONDRIAL RESPIRATORY CHAIN DEFECTS AND SYNTHETIC LETHALITY
- Given the high mutational burden of oncogenes such as RET/PTC and BRAFV600E, most differentiated and undifferentiated thyroid cancers carry genetic mutations that provide a therapeutic target. Targeted therapy for BRAFV600E and RET/PTC may not be a successful treatment for patients with radioiodine-refractory and undifferentiated thyroid cancers. Therefore, development of new anticancer drugs based on the concept of SL should facilitate identification of new therapeutic targets, thereby enabling development of new and effective treatment regimens. Moreover, identification of new SL pairs has the potential to drive drug discovery and enable rational testing of drug combinations, with the possibility of translation to thyroid cancer. Translation to the clinic is likely to require collaboration to advance the application of SL drugs to thyroid cancer.
- Current areas of interest include exploiting the DNA damage response, metabolic reprogramming, and aberrant tyrosine kinase signaling pathways. At present, relatively few SL pairs have been identified and conclusively validated for thyroid cancer. Furthermore, advances in genome engineering through siRNA and CRISPR/Cas9 technology will provide an effective platform for large-scale screens to identify new SL partners. Future discoveries in the area of SL promise to unveil new therapeutic avenues for effective and personalized treatment of thyroid cancer.
CONCLUSIONS
-
Acknowledgements
- This research was supported by a grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health and Welfare, Republic of Korea (Grant number: HR20C0025).
-
CONFLICTS OF INTEREST
No potential conflict of interest relevant to this article was reported.
Article information
Fig. 1The principles of synthetic lethality (SL) in cancer. Loss or inhibition of either of the protein products of gene A or B alone, or overexpression of gene A, is viable. However, pharmacological intervention by the partner gene product will result in an SL interaction in tumor cells with a loss-of-function mutation in a tumor suppressor gene (TSG) (A). In addition, pharmacological intervention by the partner gene product will result in a synthetic dosage lethality (SDL) interaction in tumor cells with a gain-of-function mutation or overexpression of the oncogene (B). The yellow star denotes a mutation. The thicker arrow denotes overexpression. The crossed line denotes inhibition of the gene product by pharmacological intervention.
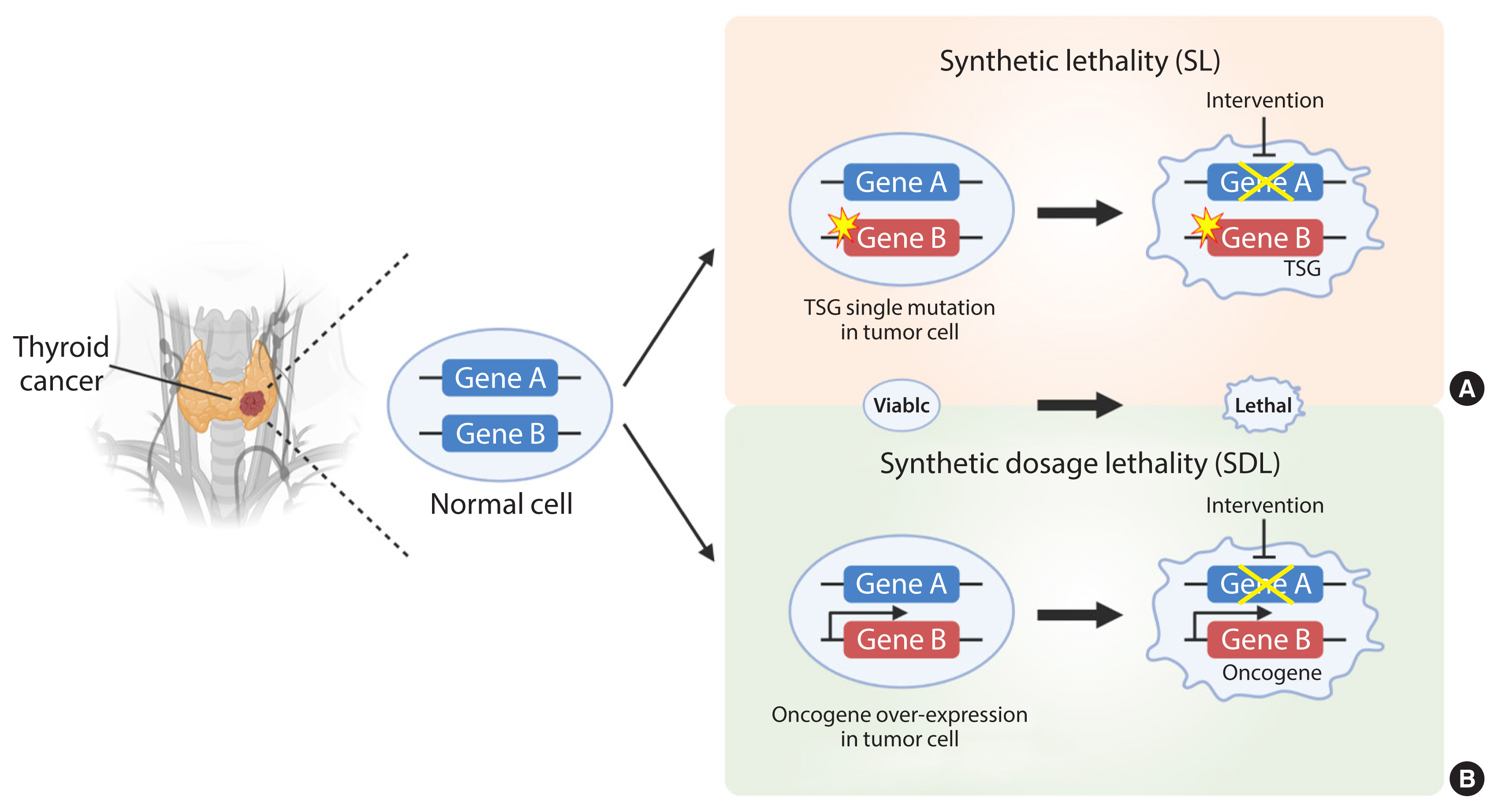
Fig. 2Application of metabolic synthetic lethality (SL) to cancer. The main metabolic pathways involved in metabolic reprogramming of cancer cells harboring mutant genes that may provide a target for SL.
- 1. Bridges CB. The origin of variations in sexual and sex-limited characters. Am Nat 1922;56:51–63.Article
- 2. Zhang B, Tang C, Yao Y, Chen X, Zhou C, Wei Z, et al. The tumor therapy landscape of synthetic lethality. Nat Commun 2021;12:1275.ArticlePubMedPMC
- 3. Ashworth A, Lord CJ. Synthetic lethal therapies for cancer: what’s next after PARP inhibitors? Nat Rev Clin Oncol 2018;15:564–76.ArticlePubMed
- 4. O’Neil NJ, Bailey ML, Hieter P. Synthetic lethality and cancer. Nat Rev Genet 2017;18:613–23.ArticlePubMed
- 5. Chen M, Cai X. Synthetic lethality is a novel and potential paradigm for precision medicine in advanced hepatocellular carcinoma. Liver Cancer 2020;9:225–6.ArticlePubMed
- 6. Sajesh BV, Guppy BJ, McManus KJ. Synthetic genetic targeting of genome instability in cancer. Cancers (Basel) 2013;5:739–61.ArticlePubMedPMC
- 7. Ashworth A, Lord CJ, Reis-Filho JS. Genetic interactions in cancer progression and treatment. Cell 2011;145:30–8.ArticlePubMed
- 8. Hartwell LH, Szankasi P, Roberts CJ, Murray AW, Friend SH. Integrating genetic approaches into the discovery of anticancer drugs. Science 1997;278:1064–8.ArticlePubMed
- 9. Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell 2009;136:823–37.ArticlePubMedPMC
- 10. Dhoonmoon A, Schleicher EM, Clements KE, Nicolae CM, Moldovan GL. Genome-wide CRISPR synthetic lethality screen identifies a role for the ADP-ribosyltransferase PARP14 in DNA replication dynamics controlled by ATR. Nucleic Acids Res 2020;48:7252–64.ArticlePubMedPMC
- 11. Parrish P, Thomas JD, Gabel AM, Kamlapurkar S, Bradley RK, Berger AH. Discovery of synthetic lethal and tumor suppressor paralog pairs in the human genome. Cell Rep 2021;36:109597.ArticlePubMedPMC
- 12. Haince JF, Rouleau M, Hendzel MJ, Masson JY, Poirier GG. Targeting poly(ADP-ribosyl)ation: a promising approach in cancer therapy. Trends Mol Med 2005;11:456–63.ArticlePubMed
- 13. Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005;434:917–21.ArticlePubMed
- 14. Vieri M, Preisinger C, Schemionek M, Salimi A, Patterson JB, Samali A, et al. Targeting of BCR-ABL1 and IRE1α induces synthetic lethality in Philadelphia-positive acute lymphoblastic leukemia. Carcinogenesis 2021;42:272–84.ArticlePubMed
- 15. Kim D, Hwang JH, Suh JM, Kim H, Song JH, Hwang ES, et al. RET/PTC (rearranged in transformation/papillary thyroid carcinomas) tyrosine kinase phosphorylates and activates phosphoinositide-dependent kinase 1 (PDK1): an alternative phosphatidylinositol 3-kinase-independent pathway to activate PDK1. Mol Endocrinol 2003;17:1382–94.ArticlePubMed
- 16. Wells SA Jr, Santoro M. Update: the status of clinical trials with kinase inhibitors in thyroid cancer. J Clin Endocrinol Metab 2014;99:1543–55.ArticlePubMedPMC
- 17. Eiring AM, Page B, Kraft IL, Mason CC, Vellore NA, Resetca D, et al. Combined STAT3 and BCR-ABL1 inhibition induces synthetic lethality in therapy-resistant chronic myeloid leukemia. Leukemia 2015;29:586–97.ArticlePubMed
- 18. Aguirre AJ, Hahn WC. Synthetic lethal vulnerabilities in KRAS-mutant cancers. Cold Spring Harb Perspect Med 2018;8:a031518.ArticlePubMedPMC
- 19. Hu K, Li K, Lv J, Feng J, Chen J, Wu H, et al. Suppression of the SLC7A11/glutathione axis causes synthetic lethality in KRAS-mutant lung adenocarcinoma. J Clin Invest 2020;130:1752–66.ArticlePubMedPMC
- 20. Schulten HJ, Salama S, Al-Ahmadi A, Al-Mansouri Z, Mirza Z, Al-Ghamdi K, et al. Comprehensive survey of HRAS, KRAS, and NRAS mutations in proliferative thyroid lesions from an ethnically diverse population. Anticancer Res 2013;33:4779–84.PubMed
- 21. Zhu X, Zhao L, Park JW, Willingham MC, Cheng SY. Synergistic signaling of KRAS and thyroid hormone receptor β mutants promotes undifferentiated thyroid cancer through MYC up-regulation. Neoplasia 2014;16:757–69.ArticlePubMedPMC
- 22. McKeown MR, Bradner JE. Therapeutic strategies to inhibit MYC. Cold Spring Harb Perspect Med 2014;4:a014266.ArticlePubMedPMC
- 23. Cermelli S, Jang IS, Bernard B, Grandori C. Synthetic lethal screens as a means to understand and treat MYC-driven cancers. Cold Spring Harb Perspect Med 2014;4:a014209.ArticlePubMedPMC
- 24. Kessler JD, Kahle KT, Sun T, Meerbrey KL, Schlabach MR, Schmitt EM, et al. A SUMOylation-dependent transcriptional subprogram is required for Myc-driven tumorigenesis. Science 2012;335:348–53.ArticlePubMed
- 25. Liu L, Ulbrich J, Muller J, Wustefeld T, Aeberhard L, Kress TR, et al. Deregulated MYC expression induces dependence upon AMPK-related kinase 5. Nature 2012;483:608–12.ArticlePubMed
- 26. Toyoshima M, Howie HL, Imakura M, Walsh RM, Annis JE, Chang AN, et al. Functional genomics identifies therapeutic targets for MYC-driven cancer. Proc Natl Acad Sci U S A 2012;109:9545–50.ArticlePubMedPMC
- 27. Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab 2016;23:27–47.ArticlePubMedPMC
- 28. Lee J, Chang JY, Kang YE, Yi S, Lee MH, Joung KH, et al. Mitochondrial energy metabolism and thyroid cancers. Endocrinol Metab (Seoul) 2015;30:117–23.ArticlePubMedPMC
- 29. Bajrami I, Walker C, Krastev DB, Weekes D, Song F, Wicks AJ, et al. Sirtuin inhibition is synthetic lethal with BRCA1 or BRCA2 deficiency. Commun Biol 2021;4:1270.ArticlePubMedPMC
- 30. Villanueva MT. Anticancer therapy: metabolic synthetic lethality. Nat Rev Drug Discov 2018;17:543.ArticlePubMedPDF
- 31. Molina JR, Sun Y, Protopopova M, Gera S, Bandi M, Bristow C, et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat Med 2018;24:1036–46.ArticlePubMed
- 32. Zecchini V, Frezza C. Metabolic synthetic lethality in cancer therapy. Biochim Biophys Acta Bioenerg 2017;1858:723–31.ArticlePubMed
- 33. Ban EJ, Kim D, Kim JK, Kang SW, Lee J, Jeong JJ, et al. Lactate dehydrogenase A as a potential new biomarker for thyroid cancer. Endocrinol Metab (Seoul) 2021;36:96–105.ArticlePubMedPMC
- 34. Bao L, Xu T, Lu X, Huang P, Pan Z, Ge M. Metabolic reprogramming of thyroid cancer cells and crosstalk in their microenvironment. Front Oncol 2021;11:773028.ArticlePubMedPMC
- 35. Pathria G, Scott DA, Feng Y, Sang Lee J, Fujita Y, Zhang G, et al. Targeting the Warburg effect via LDHA inhibition engages ATF4 signaling for cancer cell survival. EMBO J 2018;37:e99735.PubMedPMC
- 36. Le A, Cooper CR, Gouw AM, Dinavahi R, Maitra A, Deck LM, et al. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc Natl Acad Sci U S A 2010;107:2037–42.ArticlePubMedPMC
- 37. Scaglia N, Chisholm JW, Igal RA. Inhibition of stearoylCoA desaturase-1 inactivates acetyl-CoA carboxylase and impairs proliferation in cancer cells: role of AMPK. PLoS One 2009;4:e6812.ArticlePubMedPMC
- 38. Cheong JH, Park ES, Liang J, Dennison JB, Tsavachidou D, Nguyen-Charles C, et al. Dual inhibition of tumor energy pathway by 2-deoxyglucose and metformin is effective against a broad spectrum of preclinical cancer models. Mol Cancer Ther 2011;10:2350–62.ArticlePubMedPMC
- 39. Benjamin D, Robay D, Hindupur SK, Pohlmann J, Colombi M, El-Shemerly MY, et al. Dual inhibition of the lactate transporters MCT1 and MCT4 is synthetic lethal with metformin due to NAD+ depletion in cancer cells. Cell Rep 2018;25:3047–58.ArticlePubMedPMC
- 40. Benjamin D, Colombi M, Hindupur SK, Betz C, Lane HA, El-Shemerly MY, et al. Syrosingopine sensitizes cancer cells to killing by metformin. Sci Adv 2016;2:e1601756.ArticlePubMedPMC
- 41. Durai L, Ravindran S, Arvind K, Karunagaran D, Vijayalakshmi R. Synergistic effect of metformin and vemurufenib (PLX4032) as a molecular targeted therapy in anaplastic thyroid cancer: an in vitro study. Mol Biol Rep 2021;48:7443–56.ArticlePubMed
- 42. Yang M, Vousden KH. Serine and one-carbon metabolism in cancer. Nat Rev Cancer 2016;16:650–62.ArticlePubMed
- 43. Sun WY, Kim HM, Jung WH, Koo JS. Expression of serine/glycine metabolism-related proteins is different according to the thyroid cancer subtype. J Transl Med 2016;14:168.ArticlePubMedPMC
- 44. Favaro E, Bensaad K, Chong MG, Tennant DA, Ferguson DJ, Snell C, et al. Glucose utilization via glycogen phosphorylase sustains proliferation and prevents premature senescence in cancer cells. Cell Metab 2012;16:751–64.ArticlePubMed
- 45. Jariyal H, Weinberg F, Achreja A, Nagarath D, Srivastava A. Synthetic lethality: a step forward for personalized medicine in cancer. Drug Discov Today 2020;25:305–20.ArticlePubMed
- 46. Xing M. BRAF mutation in thyroid cancer. Endocr Relat Cancer 2005;12:245–62.ArticlePubMed
- 47. Garnett MJ, Rana S, Paterson H, Barford D, Marais R. Wild-type and mutant B-RAF activate C-RAF through distinct mechanisms involving heterodimerization. Mol Cell 2005;20:963–9.ArticlePubMed
- 48. Knauf JA, Ma X, Smith EP, Zhang L, Mitsutake N, Liao XH, et al. Targeted expression of BRAFV600E in thyroid cells of transgenic mice results in papillary thyroid cancers that undergo dedifferentiation. Cancer Res 2005;65:4238–45.ArticlePubMed
- 49. Begum S, Rosenbaum E, Henrique R, Cohen Y, Sidransky D, Westra WH. BRAF mutations in anaplastic thyroid carcinoma: implications for tumor origin, diagnosis and treatment. Mod Pathol 2004;17:1359–63.ArticlePubMed
- 50. Fallahi P, Ferrari SM, Galdiero MR, Varricchi G, Elia G, Ragusa F, et al. Molecular targets of tyrosine kinase inhibitors in thyroid cancer. Semin Cancer Biol 2022;79:180–96.ArticlePubMed
- 51. Jurchott K, Kuban RJ, Krech T, Bluthgen N, Stein U, Walther W, et al. Identification of Y-box binding protein 1 as a core regulator of MEK/ERK pathway-dependent gene signatures in colorectal cancer cells. PLoS Genet 2010;6:e1001231.ArticlePubMedPMC
- 52. Stelniec-Klotz I, Legewie S, Tchernitsa O, Witzel F, Klinger B, Sers C, et al. Reverse engineering a hierarchical regulatory network downstream of oncogenic KRAS. Mol Syst Biol 2012;8:601.ArticlePubMedPMC
- 53. Klotz-Noack K, Klinger B, Rivera M, Bublitz N, Uhlitz F, Riemer P, et al. SFPQ depletion is synthetically lethal with BRAFV600E in colorectal cancer cells. Cell Rep 2020;32:108184.ArticlePubMed
- 54. Bi O, Anene CA, Nsengimana J, Shelton M, Roberts W, Newton-Bishop J, et al. SFPQ promotes an oncogenic transcriptomic state in melanoma. Oncogene 2021;40:5192–203.ArticlePubMedPMC
- 55. Wang X, Zhang Y, Han ZG, He KY. Malignancy of cancers and synthetic lethal interactions associated with mutations of cancer driver genes. Medicine (Baltimore) 2016;95:e2697.ArticlePubMedPMC
- 56. Li Y, Su X, Feng C, Liu S, Guan H, Sun Y, et al. CYP2S1 is a synthetic lethal target in BRAFV600E-driven thyroid cancers. Signal Transduct Target Ther 2020;5:191.ArticlePubMedPMC
- 57. Kang HB, Fan J, Lin R, Elf S, Ji Q, Zhao L, et al. Metabolic rewiring by oncogenic BRAF V600E links ketogenesis pathway to BRAF-MEK1 signaling. Mol Cell 2015;59:345–58.ArticlePubMedPMC
- 58. Kaplon J, Zheng L, Meissl K, Chaneton B, Selivanov VA, Mackay G, et al. A key role for mitochondrial gatekeeper pyruvate dehydrogenase in oncogene-induced senescence. Nature 2013;498:109–12.ArticlePubMed
- 59. Scortegagna M, Lau E, Zhang T, Feng Y, Sereduk C, Yin H, et al. PDK1 and SGK3 contribute to the growth of BRAF-mutant melanomas and are potential therapeutic targets. Cancer Res 2015;75:1399–412.ArticlePubMedPMC
- 60. Porchia LM, Guerra M, Espinosa AV, Saji M, Kulp SK, Ringel MD, et al. OSU03012, a novel PDK1 inhibitor, decreases thyroid cancer proliferation and migration via multiple downstream pathways. Cancer Res 2014;66(8 Suppl):1205.
- 61. Yeh JJ, Lunetta KL, van Orsouw NJ, Moore FD Jr, Mutter GL, Vijg J, et al. Somatic mitochondrial DNA (mtDNA) mutations in papillary thyroid carcinomas and differential mtDNA sequence variants in cases with thyroid tumours. Oncogene 2000;19:2060–6.ArticlePubMed
- 62. Yi HS, Chang JY, Kim KS, Shong M. Oncogenes, mitochondrial metabolism, and quality control in differentiated thyroid cancer. Korean J Intern Med 2017;32:780–9.ArticlePubMedPMC
- 63. Raimundo N, Baysal BE, Shadel GS. Revisiting the TCA cycle: signaling to tumor formation. Trends Mol Med 2011;17:641–9.ArticlePubMedPMC
- 64. Sun Y, Bandi M, Lofton T, Smith M, Bristow CA, Carugo A, et al. Functional genomics reveals synthetic lethality between phosphogluconate dehydrogenase and oxidative phosphorylation. Cell Rep 2019;26:469–82.ArticlePubMed
References
Figure & Data
References
Citations
Citations to this article as recorded by 
- Toward Systems-Level Metabolic Analysis in Endocrine Disorders and Cancer
Aliya Lakhani, Da Hyun Kang, Yea Eun Kang, Junyoung O. Park
Endocrinology and Metabolism.2023; 38(6): 619. CrossRef - The Role of De novo Serine Biosynthesis from Glucose in Papillary Thyroid Cancer
Seong Eun Lee, Na Rae Choi, Jin-Man Kim, Mi Ae Lim, Bon Seok Koo, Yea Eun Kang
International Journal of Thyroidology.2023; 16(2): 175. CrossRef
 PubReader
PubReader ePub Link
ePub Link Cite
Cite