
Articles
- Page Path
- HOME > Endocrinol Metab > Volume 38(5); 2023 > Article
-
Review ArticleCalcium & bone metabolism Nuclear Factor-Kappa B Regulation of Osteoclastogenesis and Osteoblastogenesis
 Keypoint
Keypoint
The paper discusses the role of nuclear factor-kappa B, or NF-κB, signaling in maintaining skeletal integrity by regulating osteoclasts and osteoblasts. Cytokines like RANKL and TNF activate NF-κB, promoting osteoclast formation, while TNF receptor-associated factors, known as TRAFs, mediate varying responses. TRAF6 supports osteoclast formation, while TRAF3 inhibits it and counters osteoblast inhibition. Aging increases neutrophils expressing TGFβ and CCR5, leading to bone mass reduction and osteoporosis. The CCR5 inhibitor, maraviroc, can increase bone mass in aged mice, suggesting therapeutic potential. -
Brendan F. Boyce1,2
 , Jinbo Li1,2, Zhenqiang Yao1,2, Lianping Xing1,2
, Jinbo Li1,2, Zhenqiang Yao1,2, Lianping Xing1,2 -
Endocrinology and Metabolism 2023;38(5):504-521.
DOI: https://doi.org/10.3803/EnM.2023.501
Published online: September 26, 2023
1Department of Pathology and Laboratory Medicine, University of Rochester Medical Center, Rochester, NY, USA
2Center for Musculoskeletal Research, University of Rochester Medical Center, Rochester, NY, USA
- Corresponding author: Brendan F. Boyce Department of Pathology and Laboratory Medicine, University of Rochester Medical Center, 601 Elmwood Ave, Box 626, Rochester, NY 14642, USA Tel: +1-585-275-5837, Fax: +1-585-273-3637, E-mail: Brendan_Boyce@urmc.rochester.edu
• Received: July 14, 2023 • Revised: July 26, 2023 • Accepted: August 2, 2023
Copyright © 2023 Korean Endocrine Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- ABSTRACT
- INTRODUCTION
- NF-κB SIGNALING PATHWAYS
- ACTIVATION OF NF-κB CANONICAL SIGNALING
- ACTIVATION OF NF-κB NON-CANONICAL SIGNALING
- NF-κB SIGNALING IN OSTEOCLASTOGENESIS
- CANONICAL NF-κB SIGNALING IN OSTEOCLAST FORMATION
- NON-CANONICAL NF-κB SIGNALING IN OSTEOCLAST FORMATION
- NF-κB SIGNALING IN OSTEOBLAST FORMATION
- CANONICAL NF-κB SIGNALING IN OSTEOBLASTIC CELLS
- NON-CANONICAL NF-κB SIGNALING IN OSTEOBLASTIC CELLS
- MOLECULAR MECHANISMS BY WHICH NF-κB SIGNALING REGULATES OSTEOBLAST FORMATION AND FUNCTIONS
- ROLES FOR TRAF3 IN OSTEOBLAST FORMATION AND AGE-RELATED OSTEOPOROSIS
- CONCLUSIONS
- Article information
- References
ABSTRACT
- Maintenance of skeletal integrity requires the coordinated activity of multinucleated bone-resorbing osteoclasts and bone-forming osteoblasts. Osteoclasts form resorption lacunae on bone surfaces in response to cytokines by fusion of precursor cells. Osteoblasts are derived from mesenchymal precursors and lay down new bone in resorption lacunae during bone remodeling. Nuclear factor-kappa B (NF-κB) signaling regulates osteoclast and osteoblast formation and is activated in osteoclast precursors in response to the essential osteoclastogenic cytokine, receptor activator of NF-κB ligand (RANKL), which can also control osteoblast formation through RANK-RANKL reverse signaling in osteoblast precursors. RANKL and some pro-inflammatory cytokines, including tumor necrosis factor (TNF), activate NF-κB signaling to positively regulate osteoclast formation and functions. However, these cytokines also limit osteoclast and osteoblast formation through NF-κB signaling molecules, including TNF receptor-associated factors (TRAFs). TRAF6 mediates RANKL-induced osteoclast formation through canonical NF-κB signaling. In contrast, TRAF3 limits RANKL- and TNF-induced osteoclast formation, and it restricts transforming growth factor β (TGFβ)-induced inhibition of osteoblast formation in young and adult mice. During aging, neutrophils expressing TGFβ and C-C chemokine receptor type 5 (CCR5) increase in bone marrow of mice in response to increased NF-κB-induced CC motif chemokine ligand 5 (CCL5) expression by mesenchymal progenitor cells and injection of these neutrophils into young mice decreased bone mass. TGFβ causes degradation of TRAF3, resulting in decreased glycogen synthase kinase-3β/β-catenin-mediated osteoblast formation and age-related osteoporosis in mice. The CCR5 inhibitor, maraviroc, prevented accumulation of TGFβ+/CCR5+ neutrophils in bone marrow and increased bone mass by inhibiting bone resorption and increasing bone formation in aged mice. This paper updates current understanding of how NF-κB signaling is involved in the positive and negative regulation of cytokine-mediated osteoclast and osteoblast formation and activation with a focus on the role of TRAF3 signaling, which can be targeted therapeutically to enhance bone mass.
- Osteoclasts (OCs) are multinucleated cells that form by cytoplasmic fusion of precursors, which until recently were thought to be derived exclusively from bone marrow (BM)-derived hematopoietic stem cells (HSCs) postnatally. However, recent reports from several groups using in vivo lineage-tracing technology have significantly changed understanding of the origin and fate of OCs [1]. For example, OCs involved in bone modeling and BM cavity formation during embryonic development and in postnatal tooth eruption are derived from colony stimulating factor-1 receptor (CSF1R)-expressing erythromyeloid progenitors (EMPs) that migrate from the blood islands of the yolk sac into embryos [2]. These EMP-derived OCs also resorb bone in postnatal bone remodeling and fracture repair [3]. Studies using parabiosis and lineage-tracing reported that after skeletal development, mononuclear cells derived from HSCs not only give rise to most OCs, but some of them also fuse with pre-existing OCs derived from long-lived EMPs and thus increase the number of nuclei in these OCs [3]. Thus, EMP- and HSC-derived monocytes can fuse to form embryonic and postnatal OCs [2,3]. Some postnatal OCs are also derived from dendritic cells since deletion of tumor necrosis factor (TNF) receptor superfamily member 11a (TNFRSF11a; encodes receptor activator of nuclear factor-kappa B [NF-κB] [RANK]) in CD11c+ cells inhibited OC formation [4]. To further study the roles of RANK and CSF1R signaling in EMP- and HSC-derived OCs, Jacome-Galarza et al. [2] generated mice deficient in the genes encoding these proteins. Mice lacking either of these genes in EMPs developed osteopetrosis that resolved during the early months after birth, while mice with either gene knocked out in HSCs did not develop osteopetrosis until several months after birth, indicating a gradual shift in the source of osteoclast precursors (OCPs) [2]. Other lineage-tracing studies confirmed a hypothesis that OCs can undergo fission in a process in which mononuclear cells, called osteomorphs, can break away from multinucleated OCs and subsequently fuse with other OCs to persist as long-lasting cells [5]; these studies undermine decades of belief that OCs are short-lived cells and also that they all die with simultaneous apoptosis of all of their nuclei, which they still can do [6].
- OCPs are attracted to sites on bone surfaces destined for degradation (resorption) in response to signals derived from these sites. These signals include receptor activator of NF-κB ligand (RANKL), a multifunctional cytokine expressed by various cell types in bone and BM, including osteocytes embedded in bone matrix, stromal cells in BM, B and T lymphocytes [7] and adipocytes [8]. Bone is continually remodeled in the growing and adult skeleton in response to mechanical and other stimuli, and remodeling removes microscopic foci of damaged or worn-out bone. There are >1 million microscopic remodeling sites in the normal adult skeleton and they increase in conditions in which OC formation is increased, such as sex-steroid deficiency, inflammatory and metastatic bone disease, and hyperparathyroidism [7]. In the first three of these conditions, expression levels of pro-inflammatory cytokines, including TNF, interleukin 1 (IL-1), IL-6, and transforming growth factor β (TGFβ) are increased, and these, like parathyroid hormone, increase expression of RANKL to drive the increased osteoclastogenesis and activity [7].
- RANKL interaction with its receptor, RANK, activates NF-κB signaling in OCPs and in OCs, which secrete protons, chloride ions, and collagenases from under the specialized ruffled portion of their cell membrane that faces the bone surface to be resorbed. Hydrochloric acid forms under this ruffled border membrane and dissolves the mineral component of the bone and cathepsin K is secreted to degrade the matrix [7]. OCs move along bone surfaces in packs, enlarging resorption lacunae as they progress until the resorption process has been completed. Bone-forming osteoblasts (OBs) and osteocytes are derived from mesenchymal precursors in the BM and attach to the resorbed surface where they differentiate into OBs. Osteoblastic and osteoclastic cells communicate directly and indirectly with one another in resorption lacunae where they regulate the formation and activities of each other [9]. For example, interaction between ephrin B2, a ligand expressed on the surface of OCPs, and its receptor, ephrin type-B receptor 4 (EPHB4), on the surface of osteoblastic cells causes reverse signaling through ephrin B2 to suppress OC differentiation by inhibiting c-Fos/nuclear factor of activated T cells c1 (NFATc1) signaling [10], while forward signaling through EPHB4 into OBs promotes OB differentiation [10]. Similarly, semaphorin 3A (SEMA3A) is expressed by osteoblastic cells and inhibits bone resorption by binding to neuropilin 1 (NRP1) to inhibit RANKL-induced OC functions, and it enhances bone formation by promoting wingless (WNT)/β-catenin signaling [11]. In addition, OCs secrete vesicles containing RANK, which can bind to RANKL on OB precursors and increase bone formation through reverse signaling by activating runt-related transcription factor 2 (RUNX2) signaling [12].
- All aspects of OC formation and activation are regulated by NF-κB signaling, which also limits OC formation induced by cytokines, including RANKL and TNF [7]. In this paper, we update our review of understanding of the role of NF-κB signaling in OC formation and functions and add current understanding of its role in OB formation and functions with a focus on the roles of TNF receptor-associated factor 3 (TRAF3).
INTRODUCTION
- NF-κB is a family of transcription factors, which regulate the expression of many genes involved in inflammatory and other responses by binding to gene promoters. They were initially shown to be involved in innate and adaptive immune responses to pathogens and autoimmune stimuli and play critical roles in the initiation and maintenance of inflammatory conditions. However, they also regulate many aspects of normal cellular functions [13,14], and their activity is upregulated in many common conditions, including diabetes, atherosclerosis, and cancer [15]. The NF-κB protein family includes RelA (also known as p65), p50, p52, RelB, and c-Rel. p50 and p52 are derived from larger precursor proteins, p105, and p100, which are encoded by NF-κB1 and NF-κB2, respectively [13]. All five family members have an N-terminus Rel homology domain that allows them to form homo- and heterodimers with one another and to bind to specific DNA sequences on promoters. DNA binding requires a C-terminal transcription activation domain (TAD), which RelA, RelB, and c-Rel possess, but p50 and p52 do not; thus, p50 and p52 rely on interactions with these three other family members to positively regulate gene transcription [15]. RelA and c-Rel preferentially form heterodimers with p50, and RelA/p50 activate most of the critical signaling in the canonical pathway, which occurs within minutes after activation is initiated and lasts for 30 to 60 minutes. In OCs and many other cells, this occurs in response to cytokines, including RANKL, TNF, and IL-1, and is transient [16]. The non-canonical NF-κB pathway is activated several hours after canonical signaling has begun by translocation of RelB/p52 heterodimers to nuclei. Non-canonical signaling lasts for many hours and occurs efficiently in response to RANKL, but not to TNF in OCPs [17].
- NF-κB signaling comprises several activation steps that require ubiquitination and proteasomal degradation or processing of proteins that function as inhibitors of signaling in cells under basal/unstimulated conditions by retaining inhibitory NF-κB dimers in the cytoplasm of unstimulated cells. These inhibitory NF-κB proteins are called IκBs and include the canonical IκBs: IκBα, IκBβ, and IκBε [18]. These have multiple ankyrin repeats that allow them to bind to NF-κB dimers and interfere with the function of their nuclear localization signals. RelA/p50 heterodimers are held in an inactivate state in the cytoplasm mainly by their interaction with IκBα, but they can also bind to IκBβ. RelA:RelA homodimers and c-Rel/RelA heterodimers preferentially bind to IκBε [15]. The C-terminal portions of p105, called IκBγ, and of p100, called IκBδ, also contain multiple ankyrin repeats, which give them IκB-like functions [19,20]. The IκBγ portion of p105 binds to RelA and c-Rel, retaining them in the cytoplasm, but it can also bind to p50 molecules in RelA/p50 heterodimers [15]. Proteasomal processing of p105 occurs constitutively in unstimulated cells and excises the C-terminal portion to generate p50 [21]. Stimulation by cytokines, such as RANKL and TNF, causes phosphorylation of p105 and its rapid degradation in the proteasome without release of p50. In addition, IκBα, which binds to RelA/p50 heterodimers, is phosphorylated and degraded, which allows the translocation of existing p50/RelA protein heterodimers from the cytoplasm to nuclei. p100 also functions as an inhibitory protein in unstimulated cells when it is bound to RelB. Non-canonical NF-κB signaling causes p100 ubiquitination, but rather than degradation, it undergoes processing in the proteasome to p52, which forms RelB:p52 heterodimers that translocate to nuclei. Interestingly, RelB also can function as an IκB and can bind to RelA to prevent it from activating canonical signaling [22,23].
NF-κB SIGNALING PATHWAYS
- In response to cytokines, such as TNF and RANKL, canonical signaling is activated by a trimeric IκB kinase (IKK) complex, which consists of two catalytic subunits (IKKα and IKKβ) and a regulatory subunit, IKKγ, also called NF-κB essential modulator (NEMO) [15,23]. This IKK complex phosphorylates IκBα, leading to its polyubiquitination and degradation by the 26S proteasome and nuclear translocation of RelA:p50 dimers (Fig. 1) [15]. Most of the IKK activity in the canonical pathway in cells, including TNF- and RANKL-induced signaling in OCPs, is mediated by IKKβ. Two important additional inhibitory effects of canonical IKK signaling are that it up-regulates: (1) early expression of IκBα, which initiates a negative feedback loop and limits subsequent RelA/p50 translocation [15]; and (2) p100 expression later to limit signaling in the non-canonical pathway (Fig. 1) [24]. As will be discussed later, p100 has important inhibitory effects to limit RANKL- and TNF-induced osteoclastogenesis.
ACTIVATION OF NF-κB CANONICAL SIGNALING
- Non-canonical signaling is activated by IKKα following its phosphorylation by NF-κB-inducing kinase (NIK). However, in unstimulated cells, NIK is constitutively ubiquitinated on receptors, such as CD40 and RANK, by TRAF3 in a complex that includes TRAF2 and the inhibitor of apoptosis (IAP) proteins, cIAP1 and 2 [25]. Following stimulation by RANKL in OCPs, cIAP1/2 and TRAF2 ubiquitinate TRAF3, leading to its degradation in lysosomes and subsequent release of NIK from this complex. NIK then phosphorylates IKKα resulting in proteasomal processing of p100 to p52 and the formation of RelB:p52 heterodimers (Fig. 1) [26-28]. The IκBδ portion of p100 binds preferentially to RelB to retain it in the cytoplasm of unstimulated cells, but interestingly it also binds to and regulates RelA homodimers [29].
- As will be seen later, canonical [30] and non-canonical [31] NF-κB signaling negatively regulates mesenchymal progenitor cell (MPC) differentiation into OBs, suggesting that NF-κB inhibitors should be able to stimulate bone formation. In addition, NF-κB is involved in certain aspects of endochondral ossification during skeletogenesis [32].
ACTIVATION OF NF-κB NON-CANONICAL SIGNALING
- A role for NF-κB in bone cells was discovered unexpectedly in the mid-1990s when two groups independently generated NF-κB1/2 double knockout (dKO) mice to determine if they would have more than the modest immune deficiencies that had been detected in mice lacking NF-κB 1 or 2, RelB, or c-Rel. p65-/-mice, in contrast, died during embryogenesis from TNF-induced liver cell apoptosis [33]. These dKO mice had no tooth eruption and developed osteopetrosis because they did not form OCs [34,35]; they also had marked B cell and T cell differentiation defects and had no lymph nodes [34,35]. The osteopetrosis and immune deficiencies were reversed by wild-type (WT) mouse hematopoietic cell transplantation, indicating that the defects were not mesenchymal cell in origin [34]. These same defects were found later in RANKL and RANK knockout (KO) mice [16] in which the roles of RANKL/RANK signaling in OCs were also largely unexpected [36-38]. The NF-κB dKO and RANKL KO mice had increased numbers of RANK+ and tartrate-resistant acid phosphatase (TRAP)-, cathepsin K-, and calcitonin receptor- OCPs in their spleens [39], indicating that RANKL/NF-κB signaling is required for terminal differentiation of RANK-positive cells. The osteoclastogenic cytokines, RANKL, TNF, IL-1, and IL-6, did not rescue the OCP differentiation defect in dKO mice, indicating a central role for NF-κB in OC formation [40]. RANK expression in myeloid precursors is promoted by CSF1, which also is required for OC formation and regulates many aspects of OC formation and survival [41,42].
- NF-κB p100-/- and p105-/- mice have normal OC numbers and function in vivo, and IL-1 induces similar numbers of OCs from OCPs from these mice as it does from WT mice [40]. Mice with RelA KO only in hematopoietic cells have a defective response to RANKL in vivo [43]; and inhibition of RelA nuclear translocation in OCPs in vitro inhibits osteoclastogenesis [43], indicating an important role for RelA in OC formation. OC numbers are near normal in RelB-/- mice, but their OCPs have an impaired response to RANKL in vitro [44]. No bone phenotypes have been reported in c-Rel-/- mice, and OCPs from these mice form normal numbers of OCs in response to RANKL in vitro [45].
NF-κB SIGNALING IN OSTEOCLASTOGENESIS
- RANKL rapidly activates canonical NF-κB signaling in OCPs, resulting in a transient increase in RelA and p50 mRNA levels within an hour [46] and their recruitment to the NFATc1 promoter along with NFATc2, which, unlike NFATc1, is not required for OC formation; this induces transient auto-amplification of NFATc1 expression [47]. The main role of NFATc1 at this early stage of osteoclastogenesis appears to be to down-regulate expression of constitutively active repressors of RANK signaling [48], rather than to induce expression of osteoclastogenic genes, which requires expression of NF-κB. These repressors include Bcl6, which binds to the NFATc1 promoter in unstimulated OCPs to inhibit osteoclastogenesis; upon RANKL stimulation, NFATc1 replaces Bcl6 and facilitates NFATc1 auto-amplification. Interferon regulatory factor-8 (IRF-8), Eos, and v-maf musculoaponeurotic fibrosarcoma oncogene family protein B are additional constitutively-expressed transcriptional repressors of RANK signaling in OCPs [48]. As will be seen later, negative regulation of signaling pathways in response to osteoclastogenic cytokines can play significant roles to limit OC formation and bone loss. The major role of RelA in OC formation appears to be prevention of RANKL-induced OCP apoptosis, which is mediated by c-Jun N-terminal kinase (JNK), Bid, and caspase 3 [43].
- Following the transient increase in NFATc1 expression induced by RANKL, c-Fos and p52 levels increase in OCPs after approximately 2 hours, and these remain increased through the later stages of osteoclastogenesis without any further change in RelA or p50 mRNA levels [49]. NFATc1 expression levels don’t increase again until approximately 72 to 96 hours after RANKL treatment when they induce expression of dendritic cell-specific transmembrane protein (DC-STAMP), cathepsin K, TRAP, and other genes involved in OC resorptive functions. This increase in NFATc1 requires NF-κB-induced c-Fos expression because over-expression of c-Fos in NF-κB dKO OCPs induces NFATc1 expression and OC formation without RANKL treatment [49]. Bcl6 also binds to the DC-STAMP and cathepsin K promoters in unstimulated OCPs and presumably is removed from these sites to facilitate NFATc1-induced OCP fusion and OC activation [50]. It is not known if c-Fos expression is also required for the early transient induction of NFATc1 expression or why a more sustained expression of NFATc1 is not required to keep the constitutively active repressors of RANK signaling inactive.
- RANK recruits several molecules, including the multifunctional adaptor proteins, TRAFs 1, 2, 3, 5, and 6, and kinases, such as TGFβ-activated kinase-1 (TAK1) in OCPs in response to RANKL. Of these, only TRAF6 appears to be required for OCP differentiation in the canonical NF-κB pathway [51] because TRAF6-/- mice are osteopetrotic. TAK1 activates IKKγ leading to phosphorylation and subsequent activation of IKKβ, which phosphorylates IκB. The importance of this sequential activation process is highlighted by the reports that mice with IKKβ deleted in OC lineage cells (IKKβf/f;CD11b-Cre [52,53] or IKKβf/f;Mx1-Cre mice [52,53]) have defective OC formation and osteopetrosis, and that OCPs from mice with a constitutively active IKKβ (IKKβ-SS/EE) form OCs in the absence of RANK or RANKL treatment [54]. Expression of IKKβ, but not IKKα, is required for basal osteoclastogenesis and OC survival [52,53], and administration of a NEMO-binding domain peptide, which inhibits IKKβ activation, prevents OC formation and joint inflammation and erosion in mice with inflammatory arthritis [55]. In addition, macrophages, OCPs, and immune cells deficient in IKKβ undergo apoptosis in response to TNF, an effect that is mediated by activation of JNK signaling [52,53]. Thus, increased IKKβ signaling mediates both joint inflammation and erosion in inflammatory arthritis.
CANONICAL NF-κB SIGNALING IN OSTEOCLAST FORMATION
- NIK-/-, p100-/-, and RelB-/- mice all have normal numbers of OCs and no or minimal osteopetrosis in vivo [34,40,44,56], indicating that non-canonical signaling is not required for basal OC formation. IKKα functions downstream of NIK in non-canonical signaling, and thus it is not surprising that IKKα-/- mice have normal OC numbers and bone volume. OCPs from IKKα-/- mice, like those from NIK-/- mice, do not form OCs in response to RANKL in vitro [56], but they do form OCs in response to TNF or IL-1 [53]. In addition, mice lacking both p52 and RelB [57] as well as p100/RelB dKO mice that we generated [58] have normal OC formation and bone volume. Other investigators have suggested that NIK and RelB-mediated non-canonical signaling is required for the increased osteoclastogenesis induced by metastatic cancer in bone and in inflammatory arthritis because, unlike in WT control mice, OC numbers did not increase in the affected bones of RelB-/- mice in these models [44]. However, we found that RelB-/- mice responded similarly to WT littermates and generated large numbers of OCs, associated with marked bone loss in response to daily injections of RANKL (unpublished observation). Metastatic cancer cells and tissues in inflamed joints produce several factors in addition to RANKL, some of which can inhibit OC formation. Thus, the discrepancy between the above findings might reflect differences in the balance between stimulators and inhibitors of OC formation in the RelB-/- mice with different metastatic tumors or models of inflammatory arthritis. Furthermore, RelB-/- mice have increased expression levels of several inflammatory cytokines and develop multi-organ inflammation as they age [59,60], indicating that the inflammatory milieu in the BM of these mice is abnormal.
- RANKL-induced degradation of TRAF3 in OCPs is mediated by TRAF2/cIAP1/2 in the non-canonical pathway (Fig. 1) [61,62]. This stops TRAF3-mediated constitutive degradation of NIK, resulting in proteasomal processing of p100 to p52 and subsequent p52/RelB nuclear translocation. TRAF3 is typically degraded through the ubiquitin-proteasome pathway in B cells by E3 ubiquitin ligases, such as pellino E3 ubiquitin protein ligase 1 (Peli1) [63] and the RING-between-RING (RBR)-type ubiquitin ligase RNF216, also known as Triad3A [64], while deubiquitinases, such as OTU deubiquitinase 7B (OTUD7B), inhibit TRAF3 proteolysis and limit aberrant non-canonical NF-κB activation [65]. However, in OCPs, TRAF3’s degradation is autophagosomal, rather than proteasomal, and is prevented in vitro by chloroquine [61], which raises the pH in lysosomes, and thus inhibits lysosomal enzymes that function in acidic conditions [66]. Chloroquine has been used for many decades to treat malaria. It and hydroxychloroquine are used also to dampen immune responses in inflammatory diseases, including rheumatoid arthritis and lupus erythematosis [67,68]. By preventing TRAF3 degradation, chloroquine dose-dependently inhibited RANKL-induced OC formation in vitro and prevented parathyroid hormone-induced bone resorption and ovariectomy-induced bone loss in mice in vivo [61].
- TRAF3-/- mice die within the first week or two after birth with multi-organ inflammation, associated with uncontrolled NIK activity. This was rescued by crossing the KO mice with p100-/- mice [69], consistent with p100 acting as a negative regulator of inflammatory signaling in the non-canonical pathway. This early lethality limited study of the role of TRAF3 in OC formation and bone remodeling during aging. To circumvent this hurdle, we generated mice with TRAF3 conditionally deleted in OC lineage cells. The mice developed osteoporosis before they aged as a result of increased OC formation and bone resorption [61], providing further evidence that TRAF3 is a significant negative regulator of OC formation. These studies suggest that chloroquine could be given to humans to prevent age-and sex-steroid deficiency-related osteoporosis. However, chloroquine has side-effects, which would limit its use in these diseases and thus strategies such as targeting it to bone and away from other tissues could be considered [70].
- RANKL and TNF activate NF-κB, c-Fos, and NFATc1 sequentially in vitro in OCPs in a very similar manner (Fig. 1) [16,49,62]. However, TNF induces many fewer OCs than RANKL from WT OCPs in vitro [49]. Interestingly, TNF induces similar numbers of OCs from RANK-/- and WT OCPs in vitro, but it does not induce OC formation when administered to RANK-/- mice in vivo [62], suggesting that it induces inhibitors of osteoclastogenesis in the bone microenvironment. TNF and RANKL induce expression of the inhibitory NF-κB p100 protein [17], which is processed efficiently to p52 by RANKL, but not by TNF [62]. TNF and RANKL induced similar numbers of OCs from p100-/- OCPs in vitro [62] and TNF induced the formation of numerous OCs in p100/RANK dKO and p100/RANKL dKO mice in vivo, indicating that TNF can induce OC formation in the absence of RANKL signaling when the inhibitory protein, p100, is absent [62]. In addition, TNF-transgenic mice lacking p100 developed more severe joint erosion and inflammation than TNF-transgenic mice, providing evidence that p100 limits not only TNF-induced osteoclastogenesis, but also inflammation [62]. The importance of non-canonical signaling in TNF-induced inflammatory arthritis is further supported by the finding that NIK-/- mice have decreased joint inflammation in a model of TNF-induced arthritis [56]. These studies suggest that strategies to increase TRAF3 or p100 levels or prevent their degradation in OCPs and inflammatory cells should reduce joint inflammation and erosion in patients with TNF-mediated inflammatory arthritis.
NON-CANONICAL NF-κB SIGNALING IN OSTEOCLAST FORMATION
- OBs are derived from skeletal stem cells (SSCs), which give rise to chondrocytes, OBs, and adipocytes in developing and mature bones. Lineage-tracing studies carried out over the past decade have revealed that there are distinct populations of progenitor cells in the developing and mature skeleton that give rise to OBs in the various compartments of bone. For example, it is now evident that chondrocytes within the cartilage templates that precede bone during embryogenesis can transform into OBs and BM stromal cells [71,72]. They form the earliest bone in marrow spaces, while cells on the perichondrium give rise to OBs that form cortical bone and trabecular bone in the marrow space postnatally [73]. Progenitor-enriched mesenchymal stem cell (MSC) populations marked by leptin receptor-Cre and osterix (Osx)-Cre estrogen receptor (ER) arise from perivascular cells in BM and give rise to postnatal OBs [74,75]. However, more recent studies using a homeobox A11 (Hoxa11)-CreERT2 lineage-tracing system reported that Hoxa11 lineage-marked cells give rise to all skeletal/mesenchymal cells, including OBs, chondrocytes, and adipocytes, from E13 to 1-year-old and persist as MSCs that co-express the MSC markers, platelet-derived growth factor receptor α (PDGFRα)/CD51 and leptin receptor [76]. Hox genes play important roles in patterning the embryonic skeleton and the findings in this study suggest that Hox-expressing cells are SSCs that arise from the earliest stages of skeletal development and self-renew throughout life.
- NF-κBp50/p52-/-, RANKL-/-, and RANK-/- mice all evelop marked osteopetrosis with unresorbed trabecular bone filling their BM cavities. This phenotype indicates that these proteins are not required for OB formation or function during embryonic development. Incisor teeth do not erupt in the mice, which are kept alive after weaning if they are fed softened regular mouse chow. They continue to grow, but, like other osteopetrotic mice, they are smaller than their WT littermates, consistent with slightly defective endochondral ossification and associated abnormally thickened growth plates [32].
- Although early studies using mice with global KO of NF-κB components indicate that expression of NF-κB is not required for skeleton formation, information from mouse models with genetically modified NF-κB activation status in OB lineage cells demonstrate that both the canonical and non-canonical NF-κB signaling pathways are involved in OB functions postnatally.
NF-κB SIGNALING IN OSTEOBLAST FORMATION
- Canonical NF-κB signaling is activated by a wide array of common inflammatory factors and is largely mediated by the inhibitor of κB kinase, IKKβ, and a regulatory subunit, IKKγ/NEMO, which phosphorylates IκBα, leading to its degradation and nuclear translocation of NF-κB p65/p50 heterodimers [77]. Early studies reported that activation of canonical NF-κB signaling inhibits bone formation, based on an inhibitory effect of TNF induction of p65 on OB differentiation [78-81]. The NF-κB inhibitor, S1627, which inhibits the NF-κB DNA binding activity of p65 and p50 in OBs, promotes murine calvarial defect repair and increased bone mineral density in ovariectomized mice [82], providing additional evidence that canonical NF-κB signaling negatively regulates bone formation. However, several other groups reported that TNF-induced activation of canonical NF-κB signaling in MPCs promotes their differentiation into OBs [83] through bone morphogenic protein 2 (BMP2)-mediated upregulation of Runx2 and Osx expression [84]. These findings indicate that there are complex interactions involving cytokines and canonical NF-κB signaling that can have positive or negative regulatory effects on OBs to influence bone mass, depending upon the form of stimulation and the state of osteoblastic cell differentiation. This concept is further supported by data from mice that carry genetically modified canonical NF-κB signaling components in OB lineage cells.
- Mice carrying a constitutively active form of IKKβ in type-II collagen-expressing cells have abnormal bone and cartilage development, and decreased bone formation and expression of OB and chondrocyte marker genes [85]. This mouse model was generated by crossing R26StopIKKβ constitutive activation mice with type-II collagen-Cre mice that label OB and chondrocyte precursors [85]. In contrast, mice carrying a dominant-negative mutant of IKKγ under control of the bone gamma-carboxyglutamate protein 2 promoter that has been shown to be specifically activated in mature or differentiated OBs have increased bone mass and increased bone formation after ovariectomy (Fig. 2) [30]. Thus, activation of canonical NF-κB signaling in osteoprogenitors perturbs OB and chondrocyte maturation, while inhibition of it in mature OBs increases OB functions.
- Dedifferentiation is a process in which cells lose their specialized morphology, functions, and biochemistry to initiate cell division and revert to a less differentiated state in order to re-differentiate again [86]. For OBs, dedifferentiation is a process in which mature OBs down-regulate expression of differentiation markers, upregulate pre-OB markers, and become proliferative. OB dedifferentiation does not appear to occur in mammals because OB precursors are derived from mesenchymal lineage cells and not from dedifferentiated mature OBs [87]. However, genetic lineage-tracing studies indicate that OB dedifferentiation is an important cell source for bone tissue regeneration after amputation, fracture, and skull injuries in zebrafish [88].
- Retinoic acid and NF-κB signaling regulate OB dedifferentiation in zebrafish, and retinoic acid signaling must be downregulated for OBs to dedifferentiate [89]. NF-κB signaling works upstream of retinoic acid, and it also plays an important role in OB dedifferentiation in zebrafish [90]. For example, inhibition of canonical NF-κB signaling by over-expression of an IκBα super repressor or a constitutively active form of IKKβ in OBs via the Osx-creER system in zebrafish enhances OB dedifferentiation [90]. Treatment of zebrafish with the canonical NF-κB signaling inhibitor, Bay11-7085, which inhibits IκBα phosphorylation, or SH-23, which blocks nuclear translocation of NF-κB p65, results in mature OB dedifferentiation in response to amputation [90]. OB dedifferentiation serves as a source of new OB precursors for bone regeneration in zebrafish. However, since most studies in mice indicate that NF-κB negatively regulates OB differentiation, further studies will be needed to determine why NF-κB signaling appears to have different functions in zebrafish and mammalian OBs.
CANONICAL NF-κB SIGNALING IN OSTEOBLASTIC CELLS
- Non-canonical NF-κB signaling depends on NIK and IKKα-mediated processing of the precursor p100 to p52 to allow nuclear translocation of NF-κB RelB/p52 heterodimers [91]. NIK functions as a central signaling component in this pathway, orchestrating signals from multiple stimuli and activating the downstream kinase, IKKα. This triggers phosphorylation of p100 and its partial processing to p52, subsequently leading to persistent translocation of p52/RelB heterodimers to nuclei [91].
- Mice generated to have accumulation of a non-processable form of p100 have enhanced OB differentiation [92], and mice with deletion of p100, but retaining a functional p52, have osteopenia owing to increased OC activity and impaired OB parameters [57]. RelB loss of function mice in which RelB expression is inactivated by an insertional mutation in the RelB gene have regionally increased bone mass as they age [60]. At 4 weeks old, RelB mutant mice have normal trabecular bone volume in both metaphyseal and diaphyseal regions. Mean diaphyseal bone volume is increased 2-fold in 6 to 8-week-old mice and is further increased by 4-fold in 10 to 14-week-old mice compared to WT littermates. Interestingly, mean metaphyseal bone volumes remain similar between RelB mutant and WT mice as they age. RelB mutant mice have increased bone formation rates (BFRs) and serum bone formation markers at 4 weeks old prior to a detectable increase in diaphyseal trabecular bone, and these return to WT levels at 6 to 8 weeks old. The cellular and molecular mechanism for this transient increased diaphyseal trabecular bone volume is unknown, but MPCs from RelB mutant mice grow faster and have increased OB differentiation in vitro and form more new bone in a tibial cortical defect model [31]. Since OB defects observed in these mice may be due to secondary effects of global p100 or RelB KO, it will be important to examine OB function and bone phenotype in mice with abnormal NF-κB non-canonical signaling specifically in OB lineage cells.
- To investigate the role of NIK in OBs specifically, mice carrying a constitutively active NIK allele lacking the regulatory TRAF3 binding domain (NT3) were crossed with Osx-Cre mice. The resulting Osx-Cre;NT3 mice have inactive NIK in their Osx-expressing OB precursors, elevated serum bone turnover markers, and increased cortical and trabecular bone mass and anabolic responses to mechanical loading (Fig. 2) [93]. RNA-Seq and pathway analysis of tibiae from Osx-Cre;NT3 mice revealed distinct upregulation of receptor, kinase, and growth factor activities, including Wnts, as well as a calcium-response signature [93]. These Osx-Cre;NT3 mice also developed malignant subcutaneous soft tissue tumors, starting from 6 weeks old, with 100% penetrance. Large and cystic tumor masses were detected in the submandibular region, on the trunk, perineal area, and rarely on the limbs, with a predominance in females. Development of soft tissue tumors is related to constitutive activation of the alternative NF-κB pathway, including RelB in mesenchymal lineage cells [94].
- Conditional knockout (cKO) of IKKα specifically in OBs in mice was achieved by crossing Osx-Cre mice with IKKαfl/fl mice. IKKα cKO mice have normal trabecular and cortical bone volume at basal levels or following mechanical loading (Fig. 2) [95]. However, they lose body weight as they age with marked reductions in fat mass and adipocyte size. Young IKKα cKO mice have less weight gain and improved glucose metabolism following high fat diet. Osx-Cre;TdT reporter mice have increased Osx-Cre-mediated recombination in peripheral adipocytes from 6 weeks to 18 months, indicating that the Osx-Cre driver is also active in these adipocytes. Thus, fat loss in IKKα cKO mice is most likely due to progressive deficits of IKKα in adipocytes [95].
- Men and women have markedly different bone mass. Estrogen deficiency leads to rapid bone loss, and estrogen regulates both OC and OB functions [96]. The ER interacts with NF-κB proteins [97], and mice deficient in alternative NF-κB proteins, including NIK global KO [98] and RelB loss of function mice [60], have sexual dimorphism in bone mass. For example, female, but not male NIK- or RelB-deficient mice have significantly higher bone volume and lower OC forming potential than their WT littermates [99].
- NIK is negatively regulated by cIAPs, which promote NIK proteasomal degradation [100]. Thus, blockage of cIAPs activates the non-canonical NF-κB pathway by stabilizing NIK. The IAP antagonist, bivalent 6 (BV6), reduces bone volume and increases OC numbers in male, but not in female WT mice [99]. However, BV6 has a similar stimulatory effect on RANKL-mediated osteoclastogenesis from BM cells from both male and female mice, and it does not affect ovariectomy-induced bone loss or OB differentiation [99]. These data suggest that active estrogen signaling is not responsible for the sexual dimorphism effects of BV6 on bone.
NON-CANONICAL NF-κB SIGNALING IN OSTEOBLASTIC CELLS
- Although the impact of NF-κB on bone formation is profound, most mechanistic studies have reported that NF-κB mediates its action on OBs indirectly. However, recent studies have demonstrated direct roles of NF-κB signaling to regulate OB differentiation and function. Critical signaling pathways and transcription factors are involved in embryonic osteoblastogenesis, including BMPs/Runx2 and Wnt-β-catenin, which interact with both canonical and non-canonical NF-κB signaling proteins [101-103]. The BMP2 promoter contains NF-κB responsive elements, which can be activated by acetylation of p65 and p50 proteins in OBs [102]. BMPs are members of the TGFβ-superfamily, and binding of BMP to its receptor phosphorylates Smad1/5 or forms a Smad1/Smad4 complex to activate downstream signaling. NF-κB p65 associates with Smad4, but not Smad1/5 [104]. Disruption of p65 and Smad4 interactions using a site-specific peptide promotes alkaline phosphatase activity and matrix calcification by OBs induced by BMP2 in vitro and BMP-induced bone formation in vivo [104]. Runx2 is an essential transcription factor for OB differentiation and maturation, and its promoter contains two putative NF-κB binding sites to which NF-κB RelB binds directly [31]. RelB-/- MPCs have increased OB differentiation, which is inhibited by Runx2 siRNA. Thus, RelB negatively regulates OB differentiation by inhibiting Runx2 transcription/activation in MPCs [31].
- Wnt/β-catenin signaling regulates many crucial biological processes, including MPC fate and OB differentiation. In the absence of Wnt, β-catenin is phosphorylated by glycogen synthase kinase-3β (GSK3β) within a protein degradation complex. Phosphorylated β-catenin then undergoes ubiquitination and degradation in the cytosol. Wnts recruit this protein degradation complex to the cell membrane. β-Catenin cannot be phosphorylated when it is associated with GSK3β, and non-phosphorylated β-catenin accumulates in the cytosol and enters nuclei to regulate target genes [105,106]. In MPCs, NF-κB p65 promotes degradation of phosphorylated β-catenin by affecting its ubiquitination via stimulation of transcription of the ubiquitin E3 ligases, Smurf1 and Smurf2 [76].
- Osteocalcin and bone sialoprotein are also important proteins deposited in bone matrix by mature OBs. Their promoters have consensus binding sequences for NF-κB, Runx2, and β-catenin [103]. NF-κB p65/p50, but not RelB/p52, mediate stimulatory effects of Runx2 and β-catenin on osteocalcin or bone sialoprotein transcription by preventing binding of Runx2 and β-catenin to nearby binding sites. Thus, canonical NF-κB signaling directly regulates expression of bone matrix proteins [103]. Mature OBs derived from calvariae of bone gamma-carboxyglutamate protein 2/IKK dominant-negative mice in which canonical NF-κB signaling is inhibited have increased expression of Fos-related protein-1 (Fra-1), but levels of other Fos and Jun family proteins are normal in the cells [30]. Fra-1 is an essential factor for bone matrix deposition and bone formation by positively regulating transcription of bone matrix proteins [107,108].
MOLECULAR MECHANISMS BY WHICH NF-κB SIGNALING REGULATES OSTEOBLAST FORMATION AND FUNCTIONS
- Recent studies have revealed that, in addition to negatively regulating RANKL/NF-κB signaling in OCPs, TRAF3 plays important roles to maintain bone mass in the adult mouse skeleton. In addition, TRAF3 degradation by TGFβ in MPCs contributes to age-related osteoporosis [109], which is caused by a combination of increased bone resorption and reduced bone formation, associated with low-grade chronic inflammation (inflammaging) [110], fragile bones and increased risk of fractures [111]. Inflammaging is accompanied by increased production of pro-inflammatory cytokines, including TNF, IL-1, IL-6, and TGFβ [112,113], which induce increased RANKL expression, resulting in increased bone resorption and release of TGFβ from the resorbed bone matrix (Fig. 3).
- Having identified a role for TRAF3 in OCs, we investigated a possible role for TRAF3 in OB lineage cells by crossing paired related homeobox 1 (Prx1)-Cre mice with Traf3floxf/lox mice to generate Traf3f/fPrx1cre (cKO) mice, which have TRAF3 conditionally deleted in MPCs. The Prx1 gene is expressed predominantly in appendicular bones in embryos [114], but it is also expressed in developing vertebrae, and Prx1-/- mice have defects in skull, limb, and vertebral bone [115], indicating that it is more widely expressed in the skeleton than just long bones. We found that the cKO mice appear normal at birth, and at 3 months old they had normal tibial metaphyseal and vertebral trabecular bone volumes and bone formation rates (BFRs) [109]. However, at 9 months old, bone volumes at these sites were significantly lower than in control mice, associated with decreased BFRs and serum levels of the bone formation marker, osteocalcin, and increased OC numbers and surfaces and serum levels of the bone resorption marker, TRAP [109]. Therefore, this early onset osteoporosis in the TRAF3 cKO mice is a consequence of decreased bone formation and increased bone resorption, and in this regard is similar to age-related osteoporosis in WT mice [116] and humans [117]. MPCs from 9 to 12-month-old TRAF3 cKO mice had reduced differentiation potential and alkaline phosphatase colony formation, suggesting that the phenotype of the cells and reduced BFRs in the cKO mice could have been the result of long-term exposure to increasing concentrations of a factor(s), such as TGFβ, being released from bone matrix [118].
- TGFβ1 recruits MPCs [113] to support bone formation, but it also inhibits OB differentiation [119]. We speculated that TGFβ1 might mediate the inhibition of bone formation in the cKO mice and found that TGFβ1 serum levels were significantly increased in 9-month-old cKO mice [109]. TRAF3 protein levels were significantly lower in tibial metaphyseal bone of 18-than 3-month-old WT mice and in vertebral bone samples from older (53 to 80-year-old) adults than in children (8 to 18-yearold) [109]. TGFβ1 treatment increased TRAF3 ubiquitination and degradation and decreased TRAF3 protein levels in WT mouse MPCs, associated with direct binding of both cIAP1/2 and TRAF3 to TGFβ receptor I and II [120]. This binding was reduced by an IAP inhibitor, which degrades cIAP1 and cIAP2, and decreased TGFβ1-induced inhibition of OB differentiation, suggesting that cIAP1/2 and TRAF3 form a complex with TGFβRI resulting in TRAF3 ubiquitination. TRAF3 directly associated with AKT and phosphoinositide 3-kinase (PI3K), which phosphorylates β-catenin [121,122], and TGFβ1 increased this association. Our findings suggest that in young and adult mice TRAF3 forms a complex with PI3K to prevent AKT phosphorylation and degradation of β-catenin to enhance OB differentiation and maintain bone mass (Fig. 3), which is supported by our finding that TRAF3 over-expression increased OB differentiation [120].
- β-Catenin also induces osteoprotegerin (OPG) expression [123], which restricts bone resorption, while increased expression of cytokines in response to inflammaging [112,113] increases RANKL expression by osteoblastic [124] and immune cells [125], resulting in RANKL-induced TRAF3 lysosomal degradation in OCPs and NF-κB-induced increased bone resorption [109]. Consequently, TGFβ1 released in increased amounts from bone matrix during aging is activated in the acid environment in resorption lacunae and induces TRAF3 ubiquitination through cIAPs and subsequent lysosomal degradation in MPCs. This leads to activation of AKT, phosphorylation and degradation of β-catenin, inhibition of OB differentiation and increased OPG expression [123]. As a result of TRAF3 degradation in MPCs, RelA and RelB are released from their inhibitory p105 and p100 proteins and translocate to nuclei to induce RANKL expression [109], which enhances osteoclastogenesis (Fig. 3).
- Although these findings reveal important novel roles for TRAF3 in MPCs during aging, there are very small numbers of MPCs in BM, and bone resorption decreases in mice and humans during aging [116,117]. Thus, there are lower levels of TGFβ1 released from bone matrix in aged than in young mice overall, suggesting that there may be other more common sources of TGFβ1 in BM contributing to age-related osteoporosis, for example immune cells. HSCs regenerate immune and blood cells, and their numbers increase in BM during aging, when they generate more myeloid than lymphoid cells [126]. Neutrophils are candidates as another source of TGFβ1 to cause bone loss during aging. They are the most abundant sub-population of leukocytes in the blood and BM and express TGFβ1 in tumor cells [127], normal intestinal cells [128] and in respiratory cells in asthmatic subjects [129].
- Our most recent studies have identified a subset of neutrophils that express TGFβ and C-C chemokine receptor type 5 (CCR5). We called these TGFβ/CCR5-expressing neutrophils (TCNs). TCN numbers are increased in BM of 12-month-old TRAF3 cKO mice and in 18 to 22-month-old WT mice, associated with low bone mass, and they are decreased in blood, lymph nodes and spleens of these mice [120]. In contrast, 15-month-old mice with TGFβ RII conditionally deleted in MPCs (TRII-cKO mice) have low numbers of TCNs in BM and high bone mass [120]. In addition, when TCNs from aged male mice were injected into young non-obese diabetic (NOD) scid gamma (NSG) mice or implanted into NSG mice along with MPCs from WT mice they inhibited bone formation, supporting our posit that TCNs cause age-related osteoporosis [120]. We propose that during aging TCNs are the major source of TGFβ1, which induces TRAF3 degradation in BM MPCs through TGFβ receptor II signaling. This leads to increased NF-κB signaling in MPCs in BM and increased production of CCL5, which attracts more TCNs to BM, resulting in higher TGFβ1 levels and osteoporosis. Our findings support a model in which young and adult male mice have physiological levels of TGFβ, low TCN numbers in BM and restricted NF-κB-mediated production of RANKL and CCL5 by MPCs. These conditions keep bone resorption and formation at optimal levels and maintain skeletal integrity. During aging, TCNs are attracted to BM where they release increased amounts of TGFβ1, which degrades TRAF3 in MPCs, leading to increased NF-κB RelA/RelB-mediated production of RANKL and CCL5 (Fig. 3). This results in more TCNs in BM during aging, increased resorption, decreased bone formation, and osteoporosis in male mice.
- Maraviroc is an U.S. Food and Drug Administration-approved small molecule CCR5 antagonist used to treat human immunodeficiency virus (HIV)-AIDS [130]. It binds to CCR5 and blocks interaction between HIV-1 and CCR5, thus preventing HIV-1 entry into host T cells and macrophages [130]. To determine if maraviroc could have beneficial effects in osteopenic aged mice, we treated 22-month-old male WT mice with maraviroc for 4 weeks and found that it reduced TCN numbers in BM and increased vertebral bone volume, associated with increased bone formation, and decreased resorption [120]. We also gave it for 10 days to 3-month-old male NSG mice that had been injected with TCNs from aged mice and found that it had similar effects. These findings suggest that maraviroc could be a novel treatment for osteoporosis.
- These are the first reports of an important regulatory role for TRAF3 in MPCs to regulate bone mass, of RANKL expression being regulated directly by NF-κB canonical (RelA) and non-canonical (RelB) signaling, and of binding of TRAF3 to TGFβ receptor 1 to regulate downstream signaling in any cell type; they raise the possibility that TRAF3 has important negative regulatory functions in TGFβ signaling in other cell types. TRAF3 has important negative regulatory roles in immune and other cells [131], and these differ from those of other TRAFs, which typically promote NF-κB activation [132,133]. These include, TRAF3 limiting NF-κB non-canonical activation in T and B cells and inhibiting B cell survival [134], while inactivating mutations of TRAF3 are associated with human B cell lymphomas [135] and multiple myeloma [136], suggesting that TRAF3 is a tumor suppressor in B cells. In addition, mice with TRAF3 conditionally deleted in myeloid cells have early onset osteoporosis [61], multiple inflammatory diseases, infections, and tumors [137], indicating that TRAF3 is an inflammation and tumor suppressor in myeloid cells. These findings suggest that maintenance of TRAF3 levels in immune cells during aging could be a novel therapeutic approach to prevent or reduce the incidence of common age-related diseases, and not only osteoporosis.
ROLES FOR TRAF3 IN OSTEOBLAST FORMATION AND AGE-RELATED OSTEOPOROSIS
- The canonical and/or non-canonical NF-κB pathways play essential roles in certain aspects of OC, OB, and chondroblast activities, including the proliferation of precursors and the functions of differentiated cells. Some of these functions are essential during embryonic development, such as NF-κB 1 and 2 signaling in osteoclastogenesis, while others, such as RelB are restricted to activation of NF-κB signaling in OCs in disease states. NF-қB activation plays important roles in endochondral ossification to prevent dwarfism in mice and in cartilage destruction in animal models of rheumatoid arthritis and osteoarthritis, and in general negatively regulates OB formation. Although these findings suggest that drugs that promote or inhibit NF-κB activation could be developed to treat or prevent common bone diseases, only denosumab, a human monoclonal antibody, which binds to RANKL and prevents RANKL interaction with RANK and thus, indirectly, downstream NF-κB signaling, has come through clinical trials successfully to date [138]. Recent studies suggest that TRAF3 has important regulatory functions in OCs and OBs to maintain skeletal integrity in adult mice, and that during aging its degradation in OCPs by RANKL and in OB precursors by TGFβ contributes to age-related osteoporosis. Inflammaging causes increased production of RANKL by osteoblastic and immune cells in the BM and TGFβ released from resorbing bone and by TGFβ+/CCR5+ neutrophils, which are attracted to BM by CCL5-expressing mesenchymal cells are two major sources of TGFβ. TRAF3 degradation could be prevented by lysosomal inhibitors, such as chloroquine and hydroxychloroquine, and it and the CCR5 antagonist, maraviroc, could prevent the return of TGFβ+/CCL5+ neutrophils to BM. Both drugs could be repurposed to treat age-related osteoporosis.
CONCLUSIONS
-
CONFLICTS OF INTEREST
No potential conflict of interest relevant to this article was reported.
Article information
-
Acknowledgements
- Some of the research reported in this review was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health under Award Numbers R01AR43510 (to Brendan F. Boyce), AR063650 (to Lianping Xing) and by the National Institute for Aging under Award Number R01AG049994 (to Brendan F. Boyce and Zhenqiang Yao) and R01AG076731 (to Zhenqiang Yao).
Fig. 1.Canonical and non-canonical nuclear factor-kappa B (NF-κB) signaling induced by tumor necrosis factor (TNF) and receptor activator of NF-κB ligand (RANKL) in osteoclast precursors. RANKL and TNF induce canonical NF-κB signaling by recruiting TNF receptor-associated factor 6 (TRAF6) and TRAF2/5, respectively, to their receptors to activate a complex consisting of inhibitory NF-κB (IκB) kinase α (IKKα), IKKβ, and IKKγ (NF-κB essential modulator [NEMO]). This complex induces phosphorylation and degradation of IκB-α and the release of p65/p50 heterodimers, which translocate to the nucleus. p65/p50 induce expression of c-Fos and nuclear factor of activated T cells c1 (NFATc1), two other transcription factors necessary for osteoclast precursor differentiation, as well as the inhibitory κB protein, NF-κB p100. In unstimulated cells, p100 binds to RelB to prevent its translocation to the nucleus. RANKL induces the ubiquitination (Ub) and lysosomal degradation of TRAF3, releasing NF-κB-inducing kinase (NIK) to activate (phosphorylate) IKKα, which leads to proteasomal processing of p100 to p52. RelB:p52 heterodimers then go to the nucleus to induce target gene expression. TNF signaling does not degrade TRAF3, and thus NIK is degraded constitutively, leading to the accumulation of p100 in the cytoplasm of osteoclast precursors to limit their differentiation. TAK1, TGFβ-activated kinase-1; cIAP, complex inhibitor of apoptosis.
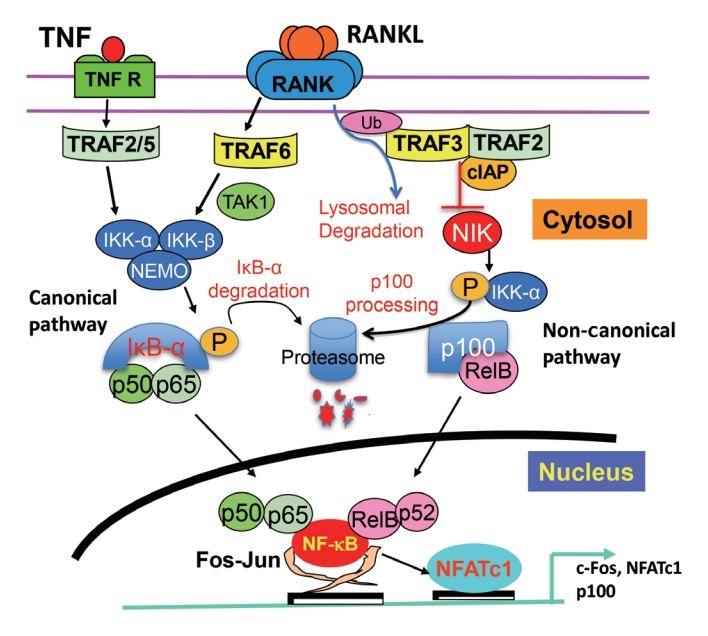
Fig. 2.Mouse models with genetically modified nuclear factor-kappa B (NF-κB) activation status in osteoblast lineage cells demonstrating that both canonical and non-canonical NF-κB signaling pathways are involved in osteoblast functions postnatally. Col2, collagen 2; IKK, inhibitory IκB kinase; OB, osteoblast; Bglap2, bone gamma-carboxyglutamate protein 2; Prx1, paired related homeobox 1; TRAF3, TNF receptor-associated factor 3; NIK, NF-κB-inducing kinase; MSC, mesenchymal stem cell; Osx, osterix.
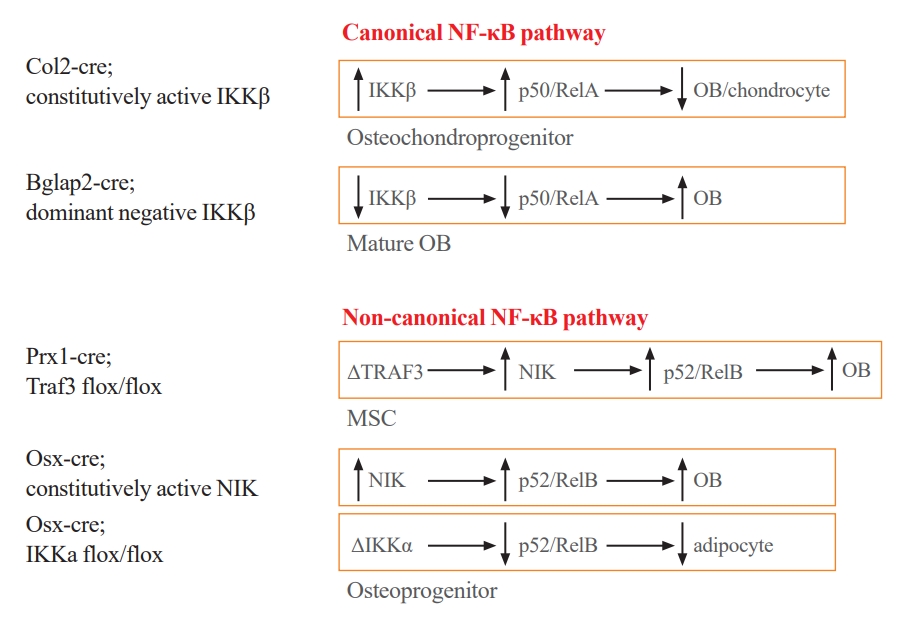
Fig. 3.Tumor necrosis factor (TNF) receptor-associated factor 3 (TRAF3) functions in osteoclast and osteoblast precursors in bone resorption and bone formation. In aged mice, increased levels of receptor activator of nuclear factor-kappa B (NF-κB) ligand (RANKL) induce (1) TRAF3 ubiquitination and subsequent lysosomal degradation in osteoclast (OC) precursors to stimulate bone resorption through NF-κB signaling. As a result, (2) transforming growth factor β1 (TGFβ1) is released from bone matrix and activated in the acid environment in resorption lacunae. Activated TGFβ1 binds to its receptor complex on mesenchymal progenitor cells (MPCs) to which TRAF3 is recruited and subsequently ubiquitinated and degraded in lysosomes. As a result, (3) NF-κB signaling is activated and both RelA and RelB bind to the RANKL promoter to further promote RANKL production, enhancing bone resorption. In young and adult mice, TRAF3 inhibits glycogen synthase kinase-3β (GSK3β) activation in MPCs to prevent β-catenin degradation, allowing β-catenin accumulation and nuclear translocation to maintain osteoblast [OB] differentiation). In aged mice, TRAF3 degradation also leads to (4) increased NF-κB-induced secretion of CC motif chemokine ligand 5 (CCL5) by MPCs, which attracts increased numbers of TGFβ1/C-C chemokine receptor type 3 (CCR3)-expressing neutrophils (TCN) to the bone marrow where they release TGFβ, leading to further degradation of TRAF3 and (5) phosphorylation of GSK3β on T216. This phosphorylation mediates degradation of β-catenin, leading to (6) inhibition of OB precursor differentiation, increased production of osteoprotegerin (OPG) and decreased bone mass.

- 1. Yahara Y, Nguyen T, Ishikawa K, Kamei K, Alman BA. The origins and roles of osteoclasts in bone development, homeostasis and repair. Development 2022;149:dev199908.ArticlePubMedPMCPDF
- 2. Jacome-Galarza CE, Percin GI, Muller JT, Mass E, Lazarov T, Eitler J, et al. Developmental origin, functional maintenance and genetic rescue of osteoclasts. Nature 2019;568:541–5.ArticlePubMedPMCPDF
- 3. Yahara Y, Barrientos T, Tang YJ, Puviindran V, Nadesan P, Zhang H, et al. Erythromyeloid progenitors give rise to a population of osteoclasts that contribute to bone homeostasis and repair. Nat Cell Biol 2020;22:49–59.ArticlePubMedPMCPDF
- 4. Tsukasaki M, Huynh NC, Okamoto K, Muro R, Terashima A, Kurikawa Y, et al. Stepwise cell fate decision pathways during osteoclastogenesis at single-cell resolution. Nat Metab 2020;2:1382–90.ArticlePubMedPDF
- 5. McDonald MM, Khoo WH, Ng PY, Xiao Y, Zamerli J, Thatcher P, et al. Osteoclasts recycle via osteomorphs during RANKL-stimulated bone resorption. Cell 2021;184:1330–47.ArticlePubMedPMC
- 6. Hughes DE, Boyce BF. Apoptosis in bone physiology and disease. Mol Pathol 1997;50:132–7.ArticlePubMedPMC
- 7. Boyce BF. Advances in the regulation of osteoclasts and osteoclast functions. J Dent Res 2013;92:860–7.ArticlePubMedPMCPDF
- 8. Yu W, Zhong L, Yao L, Wei Y, Gui T, Li Z, et al. Bone marrow adipogenic lineage precursors promote osteoclastogenesis in bone remodeling and pathologic bone loss. J Clin Invest 2021;131:e140214.ArticlePubMedPMC
- 9. Sims NA, Martin TJ. Osteoclasts provide coupling signals to osteoblast lineage cells through multiple mechanisms. Annu Rev Physiol 2020;82:507–29.ArticlePubMed
- 10. Zhao C, Irie N, Takada Y, Shimoda K, Miyamoto T, Nishiwaki T, et al. Bidirectional ephrinB2-EphB4 signaling controls bone homeostasis. Cell Metab 2006;4:111–21.ArticlePubMed
- 11. Hayashi M, Nakashima T, Taniguchi M, Kodama T, Kumanogoh A, Takayanagi H. Osteoprotection by semaphorin 3A. Nature 2012;485:69–74.ArticlePubMedPDF
- 12. Ikebuchi Y, Aoki S, Honma M, Hayashi M, Sugamori Y, Khan M, et al. Coupling of bone resorption and formation by RANKL reverse signalling. Nature 2018;561:195–200.ArticlePubMedPDF
- 13. Karin M, Greten FR. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol 2005;5:749–59.ArticlePubMedPDF
- 14. Courtois G, Gilmore TD. Mutations in the NF-kappaB signalingpathway: implications for human disease. Oncogene 2006;25:6831–43.ArticlePubMedPDF
- 15. Vallabhapurapu S, Karin M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol 2009;27:693–733.ArticlePubMed
- 16. Boyce BF, Xing L. Biology of RANK, RANKL, and osteoprotegerin. Arthritis Res Ther 2007;9(Suppl 1):S1.ArticlePubMedPMC
- 17. Novack DV, Yin L, Hagen-Stapleton A, Schreiber RD, Goeddel DV, Ross FP, et al. The IkappaB function of NF-kappaB2 p100 controls stimulated osteoclastogenesis. J Exp Med 2003;198:771–81.PubMedPMC
- 18. Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell 2008;132:344–62.ArticlePubMed
- 19. Dobrzanski P, Ryseck RP, Bravo R. Specific inhibition of RelB/p52 transcriptional activity by the C-terminal domain of p100. Oncogene 1995;10:1003–7.PubMed
- 20. Liou HC, Nolan GP, Ghosh S, Fujita T, Baltimore D. The NF-kappa B p50 precursor, p105, contains an internal I kappa B-like inhibitor that preferentially inhibits p50. EMBO J 1992;11:3003–9.ArticlePubMedPMCPDF
- 21. Palombella VJ, Rando OJ, Goldberg AL, Maniatis T. The ubiquitin-proteasome pathway is required for processing the NF-kappa B1 precursor protein and the activation of NF-kappa B. Cell 1994;78:773–85.ArticlePubMed
- 22. Overgaard M, Borch J, Gerdes K. RelB and RelE of Escherichia coli form a tight complex that represses transcription via the ribbon-helix-helix motif in RelB. J Mol Biol 2009;394:183–96.ArticlePubMedPMC
- 23. Shibata W, Maeda S, Hikiba Y, Yanai A, Ohmae T, Sakamoto K, et al. Cutting edge: the IkappaB kinase (IKK) inhibitor, NEMO-binding domain peptide, blocks inflammatory injury in murine colitis. J Immunol 2007;179:2681–5.PubMed
- 24. Chaisson ML, Branstetter DG, Derry JM, Armstrong AP, Tometsko ME, Takeda K, et al. Osteoclast differentiation is impaired in the absence of inhibitor of kappa B kinase alpha. J Biol Chem 2004;279:54841–8.PubMed
- 25. Zarnegar BJ, Wang Y, Mahoney DJ, Dempsey PW, Cheung HH, He J, et al. Noncanonical NF-kappaB activation requires coordinated assembly of a regulatory complex of the adaptors cIAP1, cIAP2, TRAF2 and TRAF3 and the kinase NIK. Nat Immunol 2008;9:1371–8.ArticlePubMedPMCPDF
- 26. Bonizzi G, Karin M. The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol 2004;25:280–8.PubMed
- 27. Senftleben U, Cao Y, Xiao G, Greten FR, Krahn G, Bonizzi G, et al. Activation by IKKalpha of a second, evolutionary conserved, NF-kappa B signaling pathway. Science 2001;293:1495–9.ArticlePubMed
- 28. Yilmaz ZB, Weih DS, Sivakumar V, Weih F. RelB is required for Peyer’s patch development: differential regulation of p52-RelB by lymphotoxin and TNF. EMBO J 2003;22:121–30.ArticlePubMedPMC
- 29. Fusco AJ, Savinova OV, Talwar R, Kearns JD, Hoffmann A, Ghosh G. Stabilization of RelB requires multidomain interactions with p100/p52. J Biol Chem 2008;283:12324–32.ArticlePubMedPMC
- 30. Chang J, Wang Z, Tang E, Fan Z, McCauley L, Franceschi R, et al. Inhibition of osteoblastic bone formation by nuclear factor-kappaB. Nat Med 2009;15:682–9.ArticlePubMedPMCPDF
- 31. Yao Z, Li Y, Yin X, Dong Y, Xing L, Boyce BF. NF-κB RelB negatively regulates osteoblast differentiation and bone formation. J Bone Miner Res 2014;29:866–77.ArticlePubMed
- 32. Xing L, Chen D, Boyce BF. Mice deficient in NF-κB p50 and p52 or RANK have defective growth plate formation and post-natal dwarfism. Bone Res 2013;1:336–45.ArticlePubMedPMC
- 33. Beg AA, Sha WC, Bronson RT, Ghosh S, Baltimore D. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-kappa B. Nature 1995;376:167–70.ArticlePubMedPDF
- 34. Franzoso G, Carlson L, Xing L, Poljak L, Shores EW, Brown KD, et al. Requirement for NF-kappaB in osteoclast and Bcell development. Genes Dev 1997;11:3482–96.ArticlePubMedPMC
- 35. Iotsova V, Caamano J, Loy J, Yang Y, Lewin A, Bravo R. Osteopetrosis in mice lacking NF-kappaB1 and NF-kappaB2. Nat Med 1997;3:1285–9.ArticlePubMedPDF
- 36. Lacey DL, Timms E, Tan HL, Kelley MJ, Dunstan CR, Burgess T, et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell 1998;93:165–76.ArticlePubMed
- 37. Yasuda H, Shima N, Nakagawa N, Yamaguchi K, Kinosaki M, Mochizuki S, et al. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc Natl Acad Sci U S A 1998;95:3597–602.ArticlePubMedPMC
- 38. Li J, Sarosi I, Yan XQ, Morony S, Capparelli C, Tan HL, et al. RANK is the intrinsic hematopoietic cell surface receptor that controls osteoclastogenesis and regulation of bone mass and calcium metabolism. Proc Natl Acad Sci U S A 2000;97:1566–71.ArticlePubMedPMC
- 39. Xing L, Bushnell TP, Carlson L, Tai Z, Tondravi M, Siebenlist U, et al. NF-kappaB p50 and p52 expression is not required for RANK-expressing osteoclast progenitor formation but is essential for RANK- and cytokine-mediated osteoclastogenesis. J Bone Miner Res 2002;17:1200–10.PubMed
- 40. Xing L, Carlson L, Story B, Tai Z, Keng P, Siebenlist U, et al. Expression of either NF-kappaB p50 or p52 in osteoclast precursors is required for IL-1-induced bone resorption. J Bone Miner Res 2003;18:260–9.PubMed
- 41. Fuller K, Owens JM, Jagger CJ, Wilson A, Moss R, Chambers TJ. Macrophage colony-stimulating factor stimulates survival and chemotactic behavior in isolated osteoclasts. J Exp Med 1993;178:1733–44.ArticlePubMedPMCPDF
- 42. Tanaka S, Takahashi N, Udagawa N, Tamura T, Akatsu T, Stanley ER, et al. Macrophage colony-stimulating factor is indispensable for both proliferation and differentiation of osteoclast progenitors. J Clin Invest 1993;91:257–63.ArticlePubMedPMC
- 43. Vaira S, Alhawagri M, Anwisye I, Kitaura H, Faccio R, Novack DV. RelA/p65 promotes osteoclast differentiation by blocking a RANKL-induced apoptotic JNK pathway in mice. J Clin Invest 2008;118:2088–97.ArticlePubMedPMC
- 44. Vaira S, Johnson T, Hirbe AC, Alhawagri M, Anwisye I, Sammut B, et al. RelB is the NF-kappaB subunit downstream of NIK responsible for osteoclast differentiation. Proc Natl Acad Sci U S A 2008;105:3897–902.PubMedPMC
- 45. Novack DV. Unique personalities within the NF-κB family: distinct functions for p65 and RelB in the osteoclast. Adv Exp Med Biol 2011;691:163–7.ArticlePubMed
- 46. Asagiri M, Sato K, Usami T, Ochi S, Nishina H, Yoshida H, et al. Autoamplification of NFATc1 expression determines its essential role in bone homeostasis. J Exp Med 2005;202:1261–9.ArticlePubMedPMCPDF
- 47. Takayanagi H, Kim S, Koga T, Nishina H, Isshiki M, Yoshida H, et al. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev Cell 2002;3:889–901.ArticlePubMed
- 48. Zhao B, Ivashkiv LB. Negative regulation of osteoclastogenesis and bone resorption by cytokines and transcriptional repressors. Arthritis Res Ther 2011;13:234.ArticlePubMedPMC
- 49. Yamashita T, Yao Z, Li F, Zhang Q, Badell IR, Schwarz EM, et al. NF-kappaB p50 and p52 regulate receptor activator of NF-kappaB ligand (RANKL) and tumor necrosis factor-induced osteoclast precursor differentiation by activating c-Fos and NFATc1. J Biol Chem 2007;282:18245–53.PubMed
- 50. Miyauchi Y, Ninomiya K, Miyamoto H, Sakamoto A, Iwasaki R, Hoshi H, et al. The Blimp1-Bcl6 axis is critical to regulate osteoclast differentiation and bone homeostasis. J Exp Med 2010;207:751–62.ArticlePubMedPMCPDF
- 51. Darnay BG, Besse A, Poblenz AT, Lamothe B, Jacoby JJ. TRAFs in RANK signaling. Adv Exp Med Biol 2007;597:152–9.ArticlePubMed
- 52. Otero JE, Dai S, Foglia D, Alhawagri M, Vacher J, Pasparakis M, et al. Defective osteoclastogenesis by IKKbeta-null precursors is a result of receptor activator of NF-kappaB ligand (RANKL)-induced JNK-dependent apoptosis and impaired differentiation. J Biol Chem 2008;283:24546–53.PubMedPMC
- 53. Ruocco MG, Maeda S, Park JM, Lawrence T, Hsu LC, Cao Y, et al. IκB kinase (IKK)β, but not IKKα, is a critical mediator of osteoclast survival and is required for inflammation-induced bone loss. J Exp Med 2005;201:1677–87.ArticlePubMedPMCPDF
- 54. Otero JE, Dai S, Alhawagri MA, Darwech I, Abu-Amer Y. IKKbeta activation is sufficient for RANK-independent osteoclast differentiation and osteolysis. J Bone Miner Res 2010;25:1282–94.ArticlePubMedPMC
- 55. Jimi E, Aoki K, Saito H, D’Acquisto F, May MJ, Nakamura I, et al. Selective inhibition of NF-kappa B blocks osteoclastogenesis and prevents inflammatory bone destruction in vivo. Nat Med 2004;10:617–24.ArticlePubMedPDF
- 56. Aya K, Alhawagri M, Hagen-Stapleton A, Kitaura H, Kanagawa O, Novack DV. NF-(kappa)B-inducing kinase controls lymphocyte and osteoclast activities in inflammatory arthritis. J Clin Invest 2005;115:1848–54.ArticlePubMedPMC
- 57. Soysa NS, Alles N, Weih D, Lovas A, Mian AH, Shimokawa H, et al. The pivotal role of the alternative NF-kappaB pathway in maintenance of basal bone homeostasis and osteoclastogenesis. J Bone Miner Res 2010;25:809–18.PubMed
- 58. Zhao C, Xiu Y, Ashton J, Xing L, Morita Y, Jordan CT, et al. Noncanonical NF-κB signaling regulates hematopoietic stem cell self-renewal and microenvironment interactions. Stem Cells 2012;30:709–18.ArticlePubMedPMCPDF
- 59. Weih F, Carrasco D, Durham SK, Barton DS, Rizzo CA, Ryseck RP, et al. Multiorgan inflammation and hematopoietic abnormalities in mice with a targeted disruption of RelB, a member of the NF-kappa B/Rel family. Cell 1995;80:331–40.ArticlePubMed
- 60. Burkly L, Hession C, Ogata L, Reilly C, Marconi LA, Olson D, et al. Expression of relB is required for the development of thymic medulla and dendritic cells. Nature 1995;373:531–6.ArticlePubMedPDF
- 61. Xiu Y, Xu H, Zhao C, Li J, Morita Y, Yao Z, et al. Chloroquine reduces osteoclastogenesis in murine osteoporosis by preventing TRAF3 degradation. J Clin Invest 2014;124:297–310.ArticlePubMed
- 62. Yao Z, Xing L, Boyce BF. NF-kappaB p100 limits TNF-induced bone resorption in mice by a TRAF3-dependent mechanism. J Clin Invest 2009;119:3024–34.ArticlePubMedPMC
- 63. Xiao Y, Jin J, Chang M, Chang JH, Hu H, Zhou X, et al. Peli1 promotes microglia-mediated CNS inflammation by regulating Traf3 degradation. Nat Med 2013;19:595–602.ArticlePubMedPMCPDF
- 64. Nakhaei P, Mesplede T, Solis M, Sun Q, Zhao T, Yang L, et al. The E3 ubiquitin ligase Triad3A negatively regulates the RIG-I/MAVS signaling pathway by targeting TRAF3 for degradation. PLoS Pathog 2009;5:e1000650.ArticlePubMedPMC
- 65. Hu H, Brittain GC, Chang JH, Puebla-Osorio N, Jin J, Zal A, et al. OTUD7B controls non-canonical NF-κB activation through deubiquitination of TRAF3. Nature 2013;494:371–4.ArticlePubMedPMCPDF
- 66. Gonzalez-Noriega A, Grubb JH, Talkad V, Sly WS. Chloroquine inhibits lysosomal enzyme pinocytosis and enhances lysosomal enzyme secretion by impairing receptor recycling. J Cell Biol 1980;85:839–52.ArticlePubMedPMCPDF
- 67. Ben-Zvi I, Kivity S, Langevitz P, Shoenfeld Y. Hydroxychloroquine: from malaria to autoimmunity. Clin Rev Allergy Immunol 2012;42:145–53.ArticlePubMedPDF
- 68. Miller AV, Ranatunga SK. Immunotherapies in rheumatologic disorders. Med Clin North Am 2012;96:475–96.ArticlePubMed
- 69. He JQ, Zarnegar B, Oganesyan G, Saha SK, Yamazaki S, Doyle SE, et al. Rescue of TRAF3-null mice by p100 NFkappa B deficiency. J Exp Med 2006;203:2413–8.PubMedPMC
- 70. Sun S, Tao J, Sedghizadeh PP, Cherian P, Junka AF, Sodagar E, et al. Bisphosphonates for delivering drugs to bone. Br J Pharmacol 2021;178:2008–25.ArticlePubMedPMCPDF
- 71. Yang G, Zhu L, Hou N, Lan Y, Wu XM, Zhou B, et al. Osteogenic fate of hypertrophic chondrocytes. Cell Res 2014;24:1266–9.ArticlePubMedPMCPDF
- 72. Yang L, Tsang KY, Tang HC, Chan D, Cheah KS. Hypertrophic chondrocytes can become osteoblasts and osteocytes in endochondral bone formation. Proc Natl Acad Sci U S A 2014;111:12097–102.ArticlePubMedPMC
- 73. Maes C, Kobayashi T, Selig MK, Torrekens S, Roth SI, Mackem S, et al. Osteoblast precursors, but not mature osteoblasts, move into developing and fractured bones along with invading blood vessels. Dev Cell 2010;19:329–44.ArticlePubMedPMC
- 74. Mizoguchi T, Pinho S, Ahmed J, Kunisaki Y, Hanoun M, Mendelson A, et al. Osterix marks distinct waves of primitive and definitive stromal progenitors during bone marrow development. Dev Cell 2014;29:340–9.ArticlePubMedPMC
- 75. Zhou BO, Yue R, Murphy MM, Peyer JG, Morrison SJ. Leptin-receptor-expressing mesenchymal stromal cells represent the main source of bone formed by adult bone marrow. Cell Stem Cell 2014;15:154–68.ArticlePubMedPMC
- 76. Pineault KM, Song JY, Kozloff KM, Lucas D, Wellik DM. Hox11 expressing regional skeletal stem cells are progenitors for osteoblasts, chondrocytes and adipocytes throughout life. Nat Commun 2019;10:3168.ArticlePubMedPMCPDF
- 77. Israel A. The IKK complex, a central regulator of NF-kappaB activation. Cold Spring Harb Perspect Biol 2010;2:a000158.PubMedPMC
- 78. Gilbert L, He X, Farmer P, Rubin J, Drissi H, van Wijnen AJ, et al. Expression of the osteoblast differentiation factor RUNX2 (Cbfa1/AML3/Pebp2alpha A) is inhibited by tumor necrosis factor-alpha. J Biol Chem 2002;277:2695–701.PubMed
- 79. Gilbert LC, Rubin J, Nanes MS. The p55 TNF receptor mediates TNF inhibition of osteoblast differentiation independently of apoptosis. Am J Physiol Endocrinol Metab 2005;288:E1011–8.ArticlePubMed
- 80. Li Y, Li A, Strait K, Zhang H, Nanes MS, Weitzmann MN. Endogenous TNFalpha lowers maximum peak bone mass and inhibits osteoblastic Smad activation through NF-kappaB. J Bone Miner Res 2007;22:646–55.ArticlePubMed
- 81. Yamazaki M, Fukushima H, Shin M, Katagiri T, Doi T, Takahashi T, et al. Tumor necrosis factor alpha represses bone morphogenetic protein (BMP) signaling by interfering with the DNA binding of Smads through the activation of NF-kappaB. J Biol Chem 2009;284:35987–95.PubMedPMC
- 82. Alles N, Soysa NS, Hayashi J, Khan M, Shimoda A, Shimokawa H, et al. Suppression of NF-kappaB increases bone formation and ameliorates osteopenia in ovariectomized mice. Endocrinology 2010;151:4626–34.PubMed
- 83. Cho HH, Shin KK, Kim YJ, Song JS, Kim JM, Bae YC, et al. NF-kappaB activation stimulates osteogenic differentiation of mesenchymal stem cells derived from human adipose tissue by increasing TAZ expression. J Cell Physiol 2010;223:168–77.PubMed
- 84. Moro GE, Minoli I, Fulconis F, Clementi M, Raiha NC. Growth and metabolic responses in low-birth-weight infants fed human milk fortified with human milk protein or with a bovine milk protein preparation. J Pediatr Gastroenterol Nutr 1991;13:150–4.ArticlePubMed
- 85. Swarnkar G, Zhang K, Mbalaviele G, Long F, Abu-Amer Y. Constitutive activation of IKK2/NF-κB impairs osteogenesis and skeletal development. PLoS One 2014;9:e91421.ArticlePubMedPMC
- 86. Tata PR, Mou H, Pardo-Saganta A, Zhao R, Prabhu M, Law BM, et al. Dedifferentiation of committed epithelial cells into stem cells in vivo. Nature 2013;503:218–23.ArticlePubMedPMCPDF
- 87. Park D, Spencer JA, Koh BI, Kobayashi T, Fujisaki J, Clemens TL, et al. Endogenous bone marrow MSCs are dynamic, fate-restricted participants in bone maintenance and regeneration. Cell Stem Cell 2012;10:259–72.ArticlePubMedPMC
- 88. Geurtzen K, Knopf F, Wehner D, Huitema LF, Schulte-Merker S, Weidinger G. Mature osteoblasts dedifferentiate in response to traumatic bone injury in the zebrafish fin and skull. Development 2014;141:2225–34.ArticlePubMedPDF
- 89. Blum N, Begemann G. Retinoic acid signaling spatially restricts osteoblasts and controls ray-interray organization during zebrafish fin regeneration. Development 2015;142:2888–93.ArticlePubMedPDF
- 90. Mishra R, Sehring I, Cederlund M, Mulaw M, Weidinger G. NF-κB signaling negatively regulates osteoblast dedifferentiation during zebrafish bone regeneration. Dev Cell 2020;52:167–82.ArticlePubMed
- 91. Sun SC. The non-canonical NF-κB pathway in immunity and inflammation. Nat Rev Immunol 2017;17:545–58.ArticlePubMedPMCPDF
- 92. Seo Y, Fukushima H, Maruyama T, Kuroishi KN, Osawa K, Nagano K, et al. Accumulation of p100, a precursor of NF-κB2, enhances osteoblastic differentiation in vitro and bone formation in vivo in aly/aly mice. Mol Endocrinol 2012;26:414–22.ArticlePubMedPMC
- 93. Davis JL, Cox L, Shao C, Lyu C, Liu S, Aurora R, et al. Conditional activation of NF-κB inducing kinase (NIK) in the osteolineage enhances both basal and loading-induced bone formation. J Bone Miner Res 2019;34:2087–100.ArticlePubMedPDF
- 94. Davis JL, Thaler R, Cox L, Ricci B, Zannit HM, Wan F, et al. Constitutive activation of NF-κB inducing kinase (NIK) in the mesenchymal lineage using Osterix (Sp7)- or Fibroblast-specific protein 1 (S100a4)-Cre drives spontaneous soft tissue sarcoma. PLoS One 2021;16:e0254426.ArticlePubMedPMC
- 95. Davis JL, Pokhrel NK, Cox L, Rohatgi N, Faccio R, Veis DJ. Conditional loss of IKKα in Osterix+cells has no effect on bone but leads to age-related loss of peripheral fat. Sci Rep 2022;12:4915.ArticlePubMedPMCPDF
- 96. Khosla S, Oursler MJ, Monroe DG. Estrogen and the skeleton. Trends Endocrinol Metab 2012;23:576–81.ArticlePubMedPMC
- 97. Biswas DK, Singh S, Shi Q, Pardee AB, Iglehart JD. Crossroads of estrogen receptor and NF-kappaB signaling. Sci STKE 2005;2005:pe27.PubMed
- 98. Yin L, Wu L, Wesche H, Arthur CD, White JM, Goeddel DV, et al. Defective lymphotoxin-beta receptor-induced NF-kappaB transcriptional activity in NIK-deficient mice. Science 2001;291:2162–5.ArticlePubMed
- 99. Zarei A, Yang C, Gibbs J, Davis JL, Ballard A, Zeng R, et al. Manipulation of the alternative NF-κB pathway in mice has sexually dimorphic effects on bone. JBMR Plus 2018;3:14–22.ArticlePubMedPMCPDF
- 100. Sun SC. Non-canonical NF-κB signaling pathway. Cell Res 2011;21:71–85.ArticlePubMedPMCPDF
- 101. Jimi E. The role of BMP signaling and NF-κB signaling on osteoblastic differentiation, cancer development, and vascular diseases: is the activation of NF-κB a friend or foe of BMP function? Vitam Horm 2015;99:145–70.ArticlePubMed
- 102. Li X, Bai XZ. NF-kappaB modulates activation of the BMP-2 gene by trichostatin A. Mol Biol (Mosk) 2008;42:990–6.PubMed
- 103. Tarapore RS, Lim J, Tian C, Pacios S, Xiao W, Reid D, et al. NF-κB has a direct role in inhibiting Bmp- and Wnt-induced matrix protein expression. J Bone Miner Res 2016;31:52–64.ArticlePubMedPDF
- 104. Urata M, Kokabu S, Matsubara T, Sugiyama G, Nakatomi C, Takeuchi H, et al. A peptide that blocks the interaction of NF-κB p65 subunit with Smad4 enhances BMP2-induced osteogenesis. J Cell Physiol 2018;233:7356–66.ArticlePubMedPDF
- 105. Baron R, Rawadi G. Targeting the Wnt/beta-catenin pathway to regulate bone formation in the adult skeleton. Endocrinology 2007;148:2635–43.ArticlePubMed
- 106. Houschyar KS, Tapking C, Borrelli MR, Popp D, Duscher D, Maan ZN, et al. Wnt pathway in bone repair and regeneration: what do we know so far. Front Cell Dev Biol 2019;6:170.ArticlePubMedPMC
- 107. Eferl R, Hoebertz A, Schilling AF, Rath M, Karreth F, Kenner L, et al. The Fos-related antigen Fra-1 is an activator of bone matrix formation. EMBO J 2004;23:2789–99.ArticlePubMedPMC
- 108. Lin X, Patil S, Gao YG, Qian A. The bone extracellular matrix in bone formation and regeneration. Front Pharmacol 2020;11:757.ArticlePubMedPMC
- 109. Li J, Ayoub A, Xiu Y, Yin X, Sanders JO, Mesfin A, et al. TGFβ-induced degradation of TRAF3 in mesenchymal progenitor cells causes age-related osteoporosis. Nat Commun 2019;10:2795.ArticlePubMedPMCPDF
- 110. Ginaldi L, Di Benedetto MC, De Martinis M. Osteoporosis, inflammation and ageing. Immun Ageing 2005;2:14.ArticlePubMedPMCPDF
- 111. Bliuc D, Nguyen ND, Milch VE, Nguyen TV, Eisman JA, Center JR. Mortality risk associated with low-trauma osteoporotic fracture and subsequent fracture in men and women. JAMA 2009;301:513–21.ArticlePubMed
- 112. Pietschmann P, Mechtcheriakova D, Meshcheryakova A, Foger-Samwald U, Ellinger I. Immunology of osteoporosis: a mini-review. Gerontology 2016;62:128–37.ArticlePubMedPMCPDF
- 113. Tang Y, Wu X, Lei W, Pang L, Wan C, Shi Z, et al. TGF-beta1-induced migration of bone mesenchymal stem cells couples bone resorption with formation. Nat Med 2009;15:757–65.ArticlePubMedPMCPDF
- 114. ten Berge D, Brouwer A, Korving J, Martin JF, Meijlink F. Prx1 and Prx2 in skeletogenesis: roles in the craniofacial region, inner ear and limbs. Development 1998;125:3831–42.ArticlePubMedPDF
- 115. Martin JF, Bradley A, Olson EN. The paired-like homeo box gene MHox is required for early events of skeletogenesis in multiple lineages. Genes Dev 1995;9:1237–49.ArticlePubMed
- 116. Ng AH, Omelon S, Variola F, Allo B, Willett TL, Alman BA, et al. Adynamic bone decreases bone toughness during aging by affecting mineral and matrix. J Bone Miner Res 2016;31:369–79.ArticlePubMedPDF
- 117. Demontiero O, Vidal C, Duque G. Aging and bone loss: new insights for the clinician. Ther Adv Musculoskelet Dis 2012;4:61–76.ArticlePubMedPMCPDF
- 118. Crane JL, Cao X. Bone marrow mesenchymal stem cells and TGF-β signaling in bone remodeling. J Clin Invest 2014;124:466–72.ArticlePubMedPMC
- 119. Lian N, Lin T, Liu W, Wang W, Li L, Sun S, et al. Transforming growth factor β suppresses osteoblast differentiation via the vimentin activating transcription factor 4 (ATF4) axis. J Biol Chem 2012;287:35975–84.ArticlePubMedPMC
- 120. Li J, Yao Z, Liu X, Duan R, Yi X, Ayoub A, et al. TGFβ1+ CCR5+ neutrophil subset increases in bone marrow and causes age-related osteoporosis in male mice. Nat Commun 2023;14:159.ArticlePubMedPMCPDF
- 121. Day TF, Guo X, Garrett-Beal L, Yang Y. Wnt/beta-catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Dev Cell 2005;8:739–50.PubMed
- 122. Hill TP, Spater D, Taketo MM, Birchmeier W, Hartmann C. Canonical Wnt/beta-catenin signaling prevents osteoblasts from differentiating into chondrocytes. Dev Cell 2005;8:727–38.PubMed
- 123. Glass DA 2nd, Bialek P, Ahn JD, Starbuck M, Patel MS, Clevers H, et al. Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev Cell 2005;8:751–64.ArticlePubMed
- 124. Udagawa N, Takahashi N, Jimi E, Matsuzaki K, Tsurukai T, Itoh K, et al. Osteoblasts/stromal cells stimulate osteoclast activation through expression of osteoclast differentiation factor/RANKL but not macrophage colony-stimulating factor: receptor activator of NF-kappa B ligand. Bone 1999;25:517–23.ArticlePubMed
- 125. Bishop KA, Coy HM, Nerenz RD, Meyer MB, Pike JW. Mouse Rankl expression is regulated in T cells by c-Fos through a cluster of distal regulatory enhancers designated the T cell control region. J Biol Chem 2011;286:20880–91.ArticlePubMedPMC
- 126. Pang WW, Price EA, Sahoo D, Beerman I, Maloney WJ, Rossi DJ, et al. Human bone marrow hematopoietic stem cells are increased in frequency and myeloid-biased with age. Proc Natl Acad Sci U S A 2011;108:20012–7.ArticlePubMedPMC
- 127. Nam JS, Terabe M, Mamura M, Kang MJ, Chae H, Stuelten C, et al. An anti-transforming growth factor beta antibody suppresses metastasis via cooperative effects on multiple cell compartments. Cancer Res 2008;68:3835–43.PubMedPMC
- 128. Chen F, Yang W, Huang X, Cao AT, Bilotta AJ, Xiao Y, et al. Neutrophils promote amphiregulin production in intestinal epithelial cells through TGF-β and contribute to intestinal homeostasis. J Immunol 2018;201:2492–501.ArticlePubMedPMCPDF
- 129. Chu HW, Trudeau JB, Balzar S, Wenzel SE. Peripheral blood and airway tissue expression of transforming growth factor beta by neutrophils in asthmatic subjects and normal control subjects. J Allergy Clin Immunol 2000;106:1115–23.PubMed
- 130. Martin-Blondel G, Brassat D, Bauer J, Lassmann H, Liblau RS. CCR5 blockade for neuroinflammatory diseases: beyond control of HIV. Nat Rev Neurol 2016;12:95–105.ArticlePubMedPDF
- 131. Sun SC. The noncanonical NF-κB pathway. Immunol Rev 2012;246:125–40.ArticlePubMedPMC
- 132. Baud V, Liu ZG, Bennett B, Suzuki N, Xia Y, Karin M. Signaling by proinflammatory cytokines: oligomerization of TRAF2 and TRAF6 is sufficient for JNK and IKK activation and target gene induction via an amino-terminal effector domain. Genes Dev 1999;13:1297–308.ArticlePubMedPMC
- 133. Shi JH, Sun SC. Tumor necrosis factor receptor-associated factor regulation of nuclear factor κB and mitogen-activated protein kinase pathways. Front Immunol 2018;9:1849.ArticlePubMedPMC
- 134. Bishop GA. TRAF3 as a powerful and multitalented regulator of lymphocyte functions. J Leukoc Biol 2016;100:919–26.ArticlePubMedPMCPDF
- 135. Nagel I, Bug S, Tonnies H, Ammerpohl O, Richter J, Vater I, et al. Biallelic inactivation of TRAF3 in a subset of B-cell lymphomas with interstitial del(14)(q24.1q32.33). Leukemia 2009;23:2153–5.ArticlePubMedPDF
- 136. Keats JJ, Fonseca R, Chesi M, Schop R, Baker A, Chng WJ, et al. Promiscuous mutations activate the noncanonical NF-kappaB pathway in multiple myeloma. Cancer Cell 2007;12:131–44.ArticlePubMedPMC
- 137. Lalani AI, Luo C, Han Y, Xie P. TRAF3: a novel tumor suppressor gene in macrophages. Macrophage (Houst) 2015;2:e1009.PubMedPMC
- 138. Burkiewicz JS, Scarpace SL, Bruce SP. Denosumab in osteoporosis and oncology. Ann Pharmacother 2009;43:1445–55.ArticlePubMedPDF
References
Figure & Data
References
Citations
Citations to this article as recorded by 
- The Role of Rosavin in the Pathophysiology of Bone Metabolism
Piotr Wojdasiewicz, Paweł Turczyn, Anna Lach-Gruba, Łukasz A. Poniatowski, Daryush Purrahman, Mohammad-Reza Mahmoudian-Sani, Dariusz Szukiewicz
International Journal of Molecular Sciences.2024; 25(4): 2117. CrossRef - The role of monocyte/macrophage chemokines in pathogenesis of osteoarthritis: A review
Hao Luo, Linfeng Li, Song Han, Tao Liu
International Journal of Immunogenetics.2024;[Epub] CrossRef - The effect of low-level laser therapy on osteoclast differentiation: Clinical implications for tooth movement and bone density
Chun-Yi Huang, Huynh Hoai Thuong Le, Hsiao-Chi Tsai, Chih-Hsin Tang, Jian-Hong Yu
Journal of Dental Sciences.2024;[Epub] CrossRef - Genetic Deficiency of the Long Pentraxin 3 Affects Osteogenesis and Osteoclastogenesis in Homeostatic and Inflammatory Conditions
Valentina Granata, Dario Strina, Maria Lucia Schiavone, Barbara Bottazzi, Alberto Mantovani, Antonio Inforzato, Cristina Sobacchi
International Journal of Molecular Sciences.2023; 24(23): 16648. CrossRef
 PubReader
PubReader ePub Link
ePub Link Cite
Cite



