Articles
- Page Path
- HOME > Endocrinol Metab > Volume 39(1); 2024 > Article
-
Namgok Lecture 2023Diabetes, obesity and metabolism Hypothalamic AMP-Activated Protein Kinase as a Whole-Body Energy Sensor and Regulator
 Keypoint
Keypoint
2023’s Namgok Lecture focuses on the role of hypothalamic AMPK, which senses the whole-body energy state. Hypothalamic AMPK activity is modulated by key metabolic hormones. Anorexigenic hormones, including leptin, insulin, and glucagon-like peptide 1, suppress hypothalamic AMPK activity, whereas the hunger hormone ghrelin activates it. Hypothalamic AMPK activity regulates energy balance and glucose homeostasis. -
Se Hee Min1*
 , Do Kyeong Song2*, Chan Hee Lee3*, Eun Roh4*, Min-Seon Kim1
, Do Kyeong Song2*, Chan Hee Lee3*, Eun Roh4*, Min-Seon Kim1 -
Endocrinology and Metabolism 2024;39(1):1-11.
DOI: https://doi.org/10.3803/EnM.2024.1922
Published online: February 14, 2024
1Division of Endocrinology and Metabolism, Department of Internal Medicine, Diabetes Center, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Korea
2Department of Internal Medicine, Ewha Womans University College of Medicine, Seoul, Korea
3Program of Material Science for Medicine and Pharmaceutics, Hallym University, Chuncheon, Korea
4Department of Internal Medicine, College of Medicine, Hallym University, Chuncheon, Korea
- Corresponding author: Min-Seon Kim. Division of Endocrinology and Metabolism, Department of Internal Medicine, Asan Medical Center, University of Ulsan College of Medicine, 88 Olympic-ro 43-gil, Songpa-gu, Seoul 05505, Korea Tel: +82-2-3010-3245, Fax: +82-2-3010-6962, E-mail: mskim@amc.seoul.kr
- *These authors contributed equally to this work.
Copyright © 2024 Korean Endocrine Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- 1,899 Views
- 74 Download
- ABSTRACT
- INTRODUCTION
- AMPK: MECHANISM OF CELLULAR ENERGY SENSING
- ROLE OF HYPOTHALAMIC AMPK IN REGULATING WHOLE-BODY ENERGY HOMEOSTASIS
- ROLE OF HYPOTHALAMIC AMPK IN REGULATING WHOLE-BODY GLUCOSE METABOLISM
- REGULATION OF HYPOTHALAMIC AMPK ACTIVITY BY METABOLIC HORMONES
- THE THERAPEUTIC POTENTIAL OF AMPK REGULATORS FOR METABOLIC DISORDERS
- CONCLUSIONS
- Article information
- References
ABSTRACT
- 5´-Adenosine monophosphate (AMP)-activated protein kinase (AMPK), a cellular energy sensor, is an essential enzyme that helps cells maintain stable energy levels during metabolic stress. The hypothalamus is pivotal in regulating energy balance within the body. Certain neurons in the hypothalamus are sensitive to fluctuations in food availability and energy stores, triggering adaptive responses to preserve systemic energy equilibrium. AMPK, expressed in these hypothalamic neurons, is instrumental in these regulatory processes. Hypothalamic AMPK activity is modulated by key metabolic hormones. Anorexigenic hormones, including leptin, insulin, and glucagon-like peptide 1, suppress hypothalamic AMPK activity, whereas the hunger hormone ghrelin activates it. These hormonal influences on hypothalamic AMPK activity are central to their roles in controlling food consumption and energy expenditure. Additionally, hypothalamic AMPK activity responds to variations in glucose concentrations. It becomes active during hypoglycemia but is deactivated when glucose is introduced directly into the hypothalamus. These shifts in AMPK activity within hypothalamic neurons are critical for maintaining glucose balance. Considering the vital function of hypothalamic AMPK in the regulation of overall energy and glucose balance, developing chemical agents that target the hypothalamus to modulate AMPK activity presents a promising therapeutic approach for metabolic conditions such as obesity and type 2 diabetes mellitus.
- Our comprehensive understanding of the mechanisms underlying body weight homeostasis and energy metabolism has significantly evolved over the last three decades. The hypothalamus plays a central role in regulating energy homeostasis [1,2]. Specific neuronal populations within this brain region respond to changes in food availability, energy reserves, and nutritional needs, thereby initiating compensatory actions that maintain whole-body energy balance [3-7]. Disruptions in the homeostatic mechanisms of energy balance have been implicated in the pathogenesis of both obesity and cachexia [5].
- 5´-Adenosine monophosphate (AMP)-activated protein kinase (AMPK) is a highly conserved serine/threonine kinase that serves as a critical regulator of cellular energy homeostasis [8,9]. AMPK monitors the cellular energy status and becomes activated in response to a decline in intracellular adenosine triphosphate (ATP) levels, which can be precipitated by metabolic stressors such as physical activity, hypoxia, or fasting [10]. Evidence suggests that AMPK modulates the whole-body energy balance, playing a crucial role in the regulation of feeding behavior and energy expenditure, primarily in the hypothalamus [11-14]. Hypothalamic AMPK also exerts a substantial influence on glucose homeostasis, which is another key aspect of homeostatic regulation by the hypothalamus [11,12].
- This review aims to provide a concise overview of the function and regulators of hypothalamic AMPK in food intake, energy expenditure, and glucose homeostasis and to discuss the therapeutic potential of targeting AMPK pathways in the treatment of metabolic diseases.
INTRODUCTION
- AMPK is a heterotrimeric complex composed of three distinct subunits: α, β, and γ. Each of these subunits plays a crucial role in the function and regulation of this enzyme. The catalytic α (α1, α2) subunit is responsible for the enzyme’s catalytic activity, while β (β1, β2) and γ (γ1, γ2, γ3) are regulatory subunits. The β subunit acts as a scaffold that holds the complex together, while the γ subunit primarily regulates AMPK activity.
- AMPK regulation is multifaceted, involving allosteric activation, phosphorylation, and dephosphorylation processes [13]. The primary mechanism for AMPK activation is the phosphorylation of Thr172 on the α subunit, which can be allosterically induced by AMP (but not adenosine diphosphate [ADP]) binding to the γ subunit [8]. This phosphorylation is predominantly carried out by upstream kinases, such as liver kinase B1 (LKB1) and Ca2+/calmodulin-dependent protein kinase kinase β (CaMKKβ) [14]. Moreover, both AMP and ADP can induce a conformational change that protects AMPK from dephosphorylation and enhances its activity [14].
- Functioning as an energy sensor, AMPK is activated in response to decreases in intracellular ATP levels and to increases in the AMP:ATP ratio, which are often the result of metabolic stressors such as exercise, hypoxia, or nutrient deprivation [8-10]. AMPK activation can also occur in the absence of a detectable increase in the AMP:ATP ratio, as seen with hyperosmolar stress [15] and during metformin treatment [16]. Once activated, AMPK phosphorylates a wide array of substrates. This action inhibits ATP-consuming anabolic pathways, including fatty acid and protein synthesis, and activates ATP-generating catabolic pathways, such as fatty acid oxidation and glycolysis [13]. The activation of AMPK generates ATP and restores the AMP:ATP and ADP:ATP ratios, thereby maintaining energy homeostasis.
AMPK: MECHANISM OF CELLULAR ENERGY SENSING
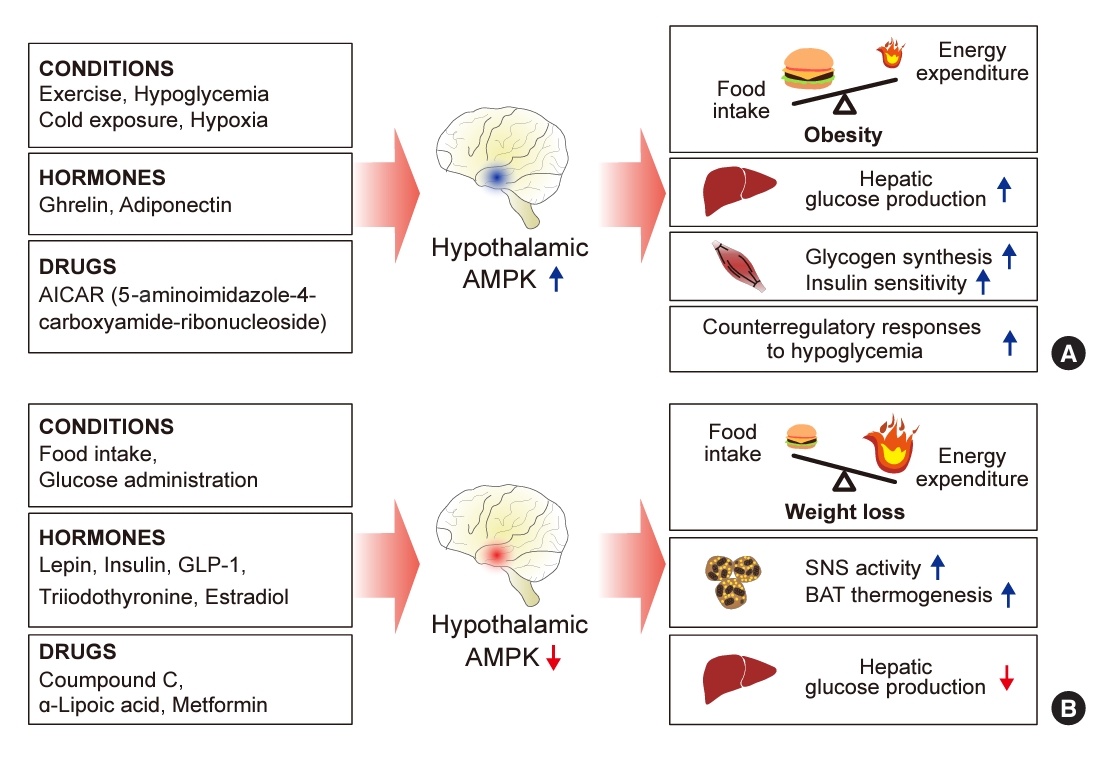
- Hypothalamic AMPK has been suggested to be a key sensor and integrator of nutritional and hormonal signals and a regulator of the whole-body energy balance (Fig. 1) [17,18]. AMPK is present in the central nervous system, with high expression levels in hypothalamic nuclei including the arcuate nucleus (ARC), dorsomedial hypothalamus, paraventricular nucleus (PVH), ventromedial nucleus (VMH), and lateral hypothalamic area (LHA). These regions are involved in controlling food intake and maintaining energy homeostasis [19].
- Alterations in hypothalamic AMPK activity influence both food intake and energy metabolism. When constitutively active (CA)-AMPK is expressed via adenovirus in the medial hypothalamus, there is an increase in food consumption and body weight. In contrast, expression of dominant-negative (DN)-AMPK in the mediobasal hypothalamus leads to reduced food intake and body weight in normal mice [20]. Similarly, activating hypothalamic AMPK by injecting the activator 5-amino4-imidazole carboxamide riboside (AICAR) into the third ventricle or the PVH results in increased food intake and body weight [21]. Conversely, reducing AMPK activity in the hypothalamus through the administration of the chemical inhibitor compound C leads to a suppression of food intake [22].
- Although the exact mechanism by which hypothalamic AMPK regulates energy homeostasis remains elusive, it is thought to mediate changes in the expression of key appetite-regulating neuropeptides in the hypothalamus, which vary according to the feeding state and the overall energy status of the body. When hypothalamic AMPK is inhibited by DN-AMPK, there is a suppression of the mRNA expression of the orexigenic neuropeptides Agouti-related peptide (AgRP) and neuropeptide Y (NPY) in the ARC during ad libitum feeding conditions. Conversely, the activation of hypothalamic AMPK by CA-AMPK expression enhances the fasting-induced increases in AgRP and NPY mRNA levels in the ARC, as well as melanin-concentrating hormone mRNA levels in the LHA [19]. Interestingly, the stimulation of AgRP and NPY mRNA expression by AMPK occurs, in part, through an autophagy-dependent mechanism [23].
- AMPK appears to regulate energy homeostasis in a hypothalamic neuron-specific manner [24,25]. When AMPK was selectively deleted from neurons expressing either proopiomelanocortin (POMC) or AgRP, both of which are pivotal for energy homeostasis, opposing effects on energy balance were observed. The deletion of the AMPKα2 catalytic subunit in POMC neurons (POMC-α2KO mice) led to obesity, characterized by increased food consumption and decreased energy expenditure. Conversely, deleting AMPKα2 in AgRP neurons (AgRP-α2KO mice) resulted in a mild, age-dependent lean phenotype, with no significant changes in food intake or energy expenditure under ad libitum feeding conditions [26]. These results imply that AMPK activity in POMC neurons may protect against obesity by reducing appetite and increasing energy expenditure, acting as a preventative mechanism. Meanwhile, the role of AMPK in AgRP neurons appears to be associated with increased adiposity through mechanisms that remain to be elucidated.
- Hypothalamic AMPK regulates energy homeostasis by modulating thermogenesis. It has been reported that several homeostatic signals target the VMH to suppress AMPK activity, which in turn promotes thermogenesis in brown adipose tissue (BAT) and the browning of white adipose tissue via the sympathetic nervous system (SNS) [20,21]. When estradiol (E2) was administered centrally, it triggered BAT thermogenesis by reducing AMPK activation in the VMH, followed by the activation of the SNS. In contrast, the genetic activation of AMPK in the VMH, using CA-AMPK adenoviruses, blocked the E2-induced enhancement of BAT thermogenesis and the associated weight loss [27]. Similarly, central administration of triiodothyronine (T3) in the VMH decreased adiposity and increased BAT thermogenesis through the SNS. However, overexpressing CA-AMPK in the VMH negated the effects of central T3 on body weight [28].
ROLE OF HYPOTHALAMIC AMPK IN REGULATING WHOLE-BODY ENERGY HOMEOSTASIS
- Hypothalamic AMPK acts as a critical sensor and integrator of whole-body glucose homeostasis, with its activity being regulated by glucose levels [25]. Intracerebroventricular (ICV) glucose administration suppresses AMPK activity in hypothalamic regions, including the ARC, PVH, and LHA [22]. In contrast, insulin-induced hypoglycemia and the inhibition of intracellular glucose utilization through 2-deoxyglucose administration lead to the activation of hypothalamic AMPK [12,22]. This activation of AMPK in response to hypoglycemia is significant in the ARC, VMH, and PVH [12].
- Several studies have suggested that hypothalamic AMPK is necessary for the hypothalamic regulation of hepatic glucose production. When conducting a pancreatic euglycemic clamp in rats, both molecular and pharmacological inhibition of hypothalamic AMPK activity led to a suppression of hepatic glucose production. This effect was mediated through the vagal motor complex pathways and the hepatic innervation of the vagus nerve [29]. Conversely, the activation of hypothalamic AMPK counteracted the glucose-lowering effects of infusing glucose or lactate directly into the hypothalamus [11].
- In addition, hypothalamic AMPK may affect whole-body insulin action through the SNS. ICV infusion of an AMPK activator, AICAR, increased insulin-stimulated muscle glycogen synthesis and insulin sensitivity when a hyperinsulinemic clamp was placed in mice [30]. Furthermore, AMPK may play a role in regulating insulin secretion through the hypothalamus. In a knockout mouse model lacking the AMPKα2 catalytic subunit gene, glucose-induced insulin secretion was found to be impaired, attributed to alterations in the autonomic nervous system [31].
- Conversely, hypothalamic AMPK also regulates the hormonal responses that counteract hypoglycemia [24]. The inhibition of hypothalamic AMPK activity with compound C diminishes these counter-regulatory responses, resulting in severe and prolonged hypoglycemia [11]. These findings indicate that hypothalamic AMPK is essential for recovery from hypoglycemia.
ROLE OF HYPOTHALAMIC AMPK IN REGULATING WHOLE-BODY GLUCOSE METABOLISM
- AMPK activity in hypothalamic neurons is regulated by important metabolic hormones, wherein AMPK acts as a downstream signaling molecule mediating their effects in the hypothalamus.
- Leptin
- Leptin, the first adipokine discovered, mediates communication between the hypothalamus and adipose tissues, which is essential for regulating food intake and energy expenditure [32]. High levels of leptin receptor expression are found in many regions of the hypothalamus [33-35]. Mutations in either the leptin receptor gene or the ob gene, which encodes leptin, lead to severe obesity in both rodents and humans, primarily due to hyperphagia [36-38]. While leptin promotes the activation of AMPK in skeletal muscle, it conversely inhibits AMPK activity in the hypothalamus [39]. Notably, the traditional leptin signaling pathway involving Janus kinase 2-signal transducer and activator of transcription 3 is not implicated in the suppression of AMPK activity [19].
- AMPK interacts with other signaling pathways, including phosphoinositide 3-kinase and the mammalian target of rapamycin complex 1, to regulate the effects of leptin in the hypothalamus [19,35,39]. Leptin stimulates POMC neurons, leading to increased production of β-endorphin and α-melanocyte-stimulating hormone by suppressing AMPK activity [36-38]. Furthermore, acetyl-CoA carboxylase (ACC), a well-established downstream target of AMPK, seems to play a role in AMPK’s influence on food intake [40]. ACC facilitates the transformation of acetyl-CoA into malonyl-CoA, while AMPK reduces ACC activity by phosphorylating it [40]. Thus, leptin-induced hypothalamic AMPK inhibition would be expected to increase malonyl-CoA levels by upregulating ACC activity [41]. Elevated malonyl-CoA levels result in increased cellular palmitoyl-CoA levels by inhibiting carnitine palmitoyl transferase-1 (CPT1) activity and subsequently suppressing mitochondrial fatty acid oxidation [41]. Increased malonyl-CoA and palmitoyl-CoA levels in hypothalamic neurons are known to suppress food intake [41]. Supporting this pathway’s role as downstream of leptin’s effects, leptin-induced inhibition of AMPK elevated malonyl-CoA levels in the ARC and palmitoyl-CoA in the PVH [39]. The pharmacological blockade of these anorexigenic fatty acids attenuated leptin’s appetite-suppressing effects.
- Insulin
- Insulin is secreted postprandially in response to various dietary cues, including glucose, fatty acids, and amino acids. It possesses lipogenic and glucose-lowering properties, and it also exerts anorexigenic effects by influencing brain activity. Animals deficient in insulin demonstrate hyperphagia [42,43]. Furthermore, mice with neuron-specific knockouts of the insulin receptor consume more food and are more susceptible to diet-induced obesity [44]. Conversely, ICV injections of insulin or its analogues produce the opposite effect [45-48].
- Insulin exerts a broad inhibitory effect on AMPKα2 activity across various regions of the hypothalamus, including the PVH, ARC, and LHA [35]. This suppression of hypothalamic AMPK by insulin is responsible for its anorexigenic effect [49]. Conversely, insulin-induced hypoglycemia increases AMPKα2 activity in the hypothalamus, a critical component of the counter-regulatory hormone response to low blood sugar levels, as previously mentioned [16]. Furthermore, insulin deficiency caused by streptozotocin leads to hyperphagia through the activation of AMPK and elevated NPY expression in the rat hypothalamus [12]. In this diabetic model, either insulin treatment or the pharmacological or molecular inhibition of AMPK in the hypothalamus can reverse hyperphagia [12], indicating that the activation of hypothalamic AMPK may be a key factor in driving hyperphagia in diabetes. The suppressive effect of insulin on hypothalamic AMPK is diminished by cold exposure, which could explain why cold conditions reduce the effectiveness of insulin in inhibiting feeding [50].
- Ghrelin
- Ghrelin is the first gastrointestinal hormone known to promote appetite. This hormone is produced in the stomach and is secreted during fasting [51]. The orexigenic effect of ghrelin is mediated by hypothalamic AgRP/NPY-producing neurons [52]. Central or peripheral administration of ghrelin in rats increases AMPK activity in the ARC and the VMH through the growth hormone secretagogue receptor expressed in these brain regions [53-56]. Ghrelin’s orexigenic effect is eliminated when hypothalamic AMPK activation is inhibited [54,57-59].
- The orexigenic effect of ghrelin seems be mediated by hypothalamic AMPK through multiple downstream targets. For example, ghrelin releases Ca2+ from the endoplasmic reticulum to activate NPY neurons via AMPK-dependent mechanisms [60-62]. Additionally, ghrelin stimulates NPY neuronal activity by activating AMPK in presynaptic neurons and by regulating synaptic plasticity in NPY neurons [36]. In another pathway, ghrelin activates NPY neurons and enhances food intake by promoting hypothalamic mitochondrial function through a mechanism dependent on uncoupling protein 2 (UCP2) [62]. Ghrelin was also found to increase UCP2 expression in hypothalamic neurons via the AMPK-ACC-CPT1-fatty acid oxidation pathway [62]. The orexigenic action of ghrelin is closely associated with fatty acid metabolism in hypothalamic VMH neurons [55]. Additionally, ghrelin downregulates fatty acid synthase in the VMH through an AMPK-dependent mechanism, which is a key factor in fasting-induced hyperphagia [55].
- Glucagon-like peptide-1
- Glucagon-like peptide-1 (GLP-1) is an incretin hormone secreted by intestinal L cells [63,64]. It is also a neuropeptide generated by preproglucagon neurons in nucleus tractus solitarius in the brainstem, which stimulates the hypothalamus to suppress appetite [63,64]. In fasted rats, the hypothalamic GLP-1 level decreased, while central GLP-1 injection inhibited food intake [65,66]. The anorectic effect of GLP-1 is achieved by its inhibitory action on hypothalamus AMPK activation [66,67]. Targeted injection of the GLP-1 receptor agonists exendin-4 or liraglutide into the VMH has been found to decrease food intake in mice and humans [68,69]. Pre-injection of AICAR in the VMH counteracted the anorexic effect of exendin-4 [69], confirming that GLP-1 induces anorexia by suppressing hypothalamic AMPK activity.
- Adiponectin
- Adiponectin, the most abundant adipokine, exerts anti-inflammatory and insulin-sensitizing effects [70,71]. People with obesity and diabetes have lower levels of circulating adiponectin than their counterparts [72,73]. Adiponectin inhibits glucose production in the liver and increases glucose uptake and fatty acid oxidation in the skeletal muscle [70,71]. In addition to its peripheral endocrine actions, adiponectin also influences food intake and energy balance through its effects on the hypothalamus. The enzyme AMPK is pivotal in mediating adiponectin’s central metabolic functions [70,71,74]. Intravenous infusion of full-length adiponectin activates AMPK in the hypothalamus, which in turn increases food consumption and reduces energy expenditure following a period of fasting [75]. The beneficial effects of adiponectin are abrogated when either DN-AMPK is overexpressed or adiponectin receptor 1 (AdipoR1) expression is silenced in the hypothalamus using adenovirus-mediated techniques or small inhibitory RNA, respectively. Additionally, the hypothalamic AdipoR1-AMPK signaling pathway is implicated in the modulation of food intake and energy expenditure by pioglitazone, a peroxisome proliferator-activated receptor γ (PPARγ) agonist [50].
REGULATION OF HYPOTHALAMIC AMPK ACTIVITY BY METABOLIC HORMONES
- Systemic AMPK stimulation is an effective approach for combating metabolic disorders, such as type 2 diabetes and obesity [76]. Activation of AMPK in adipose tissues enhances thermogenesis, fatty acid oxidation, mitochondrial function, and insulin sensitivity. This is achieved by suppressing the activity of ACC and 3-hydroxy-3-methylglutaryl coenzyme A reductase or by stimulating the phosphorylation of hormone-sensitive lipase and adipose triglyceride lipase [77,78]. In skeletal muscle, AMPK activation increases glucose uptake by promoting the translocation of glucose transporter 4 to the plasma membrane [79].
- Due to its metabolic benefits, AMPK has emerged as one of the most promising therapeutic targets for diabetes and obesity. Various drugs have been shown to enhance glucose and lipid metabolism by directly or indirectly activating AMPK signaling. Indirect activators, including biguanides (metformin), thiazolidinediones (TZDs), and α-lipoic acid (ALA), induce AMPK activation by promoting the accumulation of AMP or calcium. Direct activators, such as AICAR, thienopyridones (A-769662), and salicylate, stimulate AMPK by binding to specific subunits of the AMPK.
- Indirect AMPK activators, particularly biguanides such as metformin and phenformin, elevate cellular AMP:ATP and ADP:ATP ratios. This elevation is due to the inhibition of ATP synthesis by targeting mitochondrial electron transport chain (ETC) complex 1 [80]. As a result of decreased glycolysis and gluconeogenesis in the liver, biguanide therapy has become a mainstay in the management of type 2 diabetes [81,82]. Unlike its impact on AMPK activity in the peripheral organs, metformin inhibits AMPK activity in cultured hypothalamic neurons, thereby decreasing NPY expression and suppressing food intake [83]. Consistently, ICV administration of metformin counteracts ghrelin-induced hypothalamic AMPK activation and hyperphagia [84].
- TZDs, such as pioglitazone, rosiglitazone, and troglitazone, improve insulin sensitivity by activating PPARγ and AMPK in the skeletal muscle, liver, and adipose tissues [85,86]. TZDs activate AMPK by inhibiting the ETC complex 1, thereby increasing AMP, like biguanides [87]. Pioglitazone treatment improves insulin sensitivity and glucose tolerance by indirectly activating hypothalamic AMPK via adiponectin-AdipoR1-dependent mechanisms [50].
- ALA exerts beneficial effects on metabolic syndrome, lipotoxicity, and endothelial dysfunction through CaMKKβ-mediated intracellular calcium signaling, which is responsible for AMPK activation [88-91]. Unlike peripheral tissues, however, the central administration of ALA reduced appetite and yielded anti-obesity effects by suppressing hypothalamic AMPK activity [22]. This indicates that ALA exerts tissue-specific opposing effects on AMPK activation [22].
- Direct AMPK activators are categorized into two types: AMP mimetics and non-nucleoside activators that bind to AMPK indirectly. Unlike indirect activators, direct AMPK activators do not alter the AMP:ATP ratio or mitochondrial function, leading to the assumption that they may have fewer side effects. AICAR functions as an AMP analog; upon entering cells, it is phosphorylated by adenosine kinase to form 5-aminoimidazole-4-carboxamide ribotide (ZMP). ZMP, bearing a structural resemblance to AMP, binds to the same site on the AMPK subunit as AMP does [92]. Allosteric AMPK changes induced by ZMP promote its phosphorylation via upstream kinases such as LKB1 or CaMKKβ [93]. However, some AICAR effects are AMPK-independent [94].
- AICAR treatment has been shown to improve metabolic abnormality in mice fed a high-fat diet [95]. In human subjects, intravenous administration of AICAR reduces hepatic glucose production and inhibits lipolysis throughout the body in patients with type 2 diabetes [96]. However, although AICAR has been used clinically in cases of coronary artery disease, the therapeutic benefits have been minimal or almost negligible [97,98]. Moreover, high doses of AICAR (210 mg/kg) have been associated with renal toxicity in some patients with myelodysplastic syndrome, which suggests that caution is warranted when considering AICAR for clinical use [99].
- A-769662 has been developed as a new AMPK activator that decreases blood glucose and triglyceride levels, as well as hepatic triglyceride content [100,101]. This compound activates AMPK by binding to and phosphorylating Thr-172 in the AMPK α subunit, a key step for AMP-dependent AMPK activation [100]. Additionally, A-769662 phosphorylates Ser-108 in the AMPK α1 subunit, promoting fatty acid oxidation through phosphorylation-induced ACC inhibition [102].
- Despite the numerous beneficial effects observed in mouse models, clinical evidence supporting the use of AMPK activators for metabolic diseases remains limited. Furthermore, caution is warranted when utilizing AMPK activators because of the different metabolic outcomes observed in peripheral tissues and the brain.
THE THERAPEUTIC POTENTIAL OF AMPK REGULATORS FOR METABOLIC DISORDERS
- Evidence indicates that the regulation of AMPK activity in hypothalamic neurons is essential for maintaining whole-body energy balance and glucose homeostasis. AMPK acts as a key signaling molecule for various hormones and nutrients. Changes in AMPK activity within these neurons can significantly impact cellular signaling pathways, mitochondrial functions, autophagy, neuroelectrical properties, and transcriptional activity. These alterations are crucial for the neuronal functions that are involved in the central regulation of metabolic homeostasis.
- Although this review focuses on hypothalamic neuronal AMPK, it is important to note that AMPK in hypothalamic non-neuronal cells, such as microglia and astrocytes, may also play a role in maintaining metabolic homeostasis and preventing metabolic diseases. For instance, activation of AMPK has been shown to induce anti-inflammatory responses in microglia [103], which could help alleviate hypothalamic inflammation and obesity caused by a high-fat diet. Additionally, the anti-hypertensive drug telmisartan has been found to prevent lipopolysaccharide-induced microglial activation through CaMKKβ-induced AMPK activation [104]. In astrocytes, AMPK activation protects against hypoxia-induced cell death [105] and promotes glycolysis and lactate production via the lactate shuttle [106]. Therefore, AMPK inhibition in astrocytes causes neuronal loss [106]. To date, the role of glial AMPK in energy metabolism has not been thoroughly investigated, and the significance of AMPK in hypothalamic glial cells warrants further research.
- Given that AMPK is a key integrator and regulator of energy homeostasis, there have been ongoing trials to develop AMPK activators with beneficial metabolic effects. However, despite the identification and preclinical testing of several direct AMPK activators, only a handful have advanced to clinical trials. The results of these trials have been underwhelming, and safety concerns have emerged, especially at higher doses of AMPK activators. AMPK activation promotes positive metabolic outcomes by targeting peripheral metabolic organs. However, these benefits may be counterbalanced by AMPK activation in the brain, which could lead to increased food intake and decreased energy expenditure. Consequently, the creation of tissue-specific delivery systems for AMPK regulators may be crucial to improve metabolic outcomes and mitigate potential side effects.
CONCLUSIONS
-
CONFLICTS OF INTEREST
No potential conflict of interest relevant to this article was reported.
Article information
-
Acknowledgements
- This study was supported by grants from the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT of Korea (2020R1A2C3004843, 2022M3E5E8017213 to Min-Seon Kim, 2022R1C1C1012590 to Se Hee Min, 2022 R1C1C1004187 to Chan Hee Lee; 2022R1F1A1071743 to Do Kyeong Song).

- 1. Roh E, Kim MS. Brain regulation of energy metabolism. Endocrinol Metab (Seoul) 2016;31:519–24.ArticlePubMedPMCPDF
- 2. Roh E, Song DK, Kim MS. Emerging role of the brain in the homeostatic regulation of energy and glucose metabolism. Exp Mol Med 2016;48:e216.ArticlePubMedPMCPDF
- 3. Quarta C, Claret M, Zeltser LM, Williams KW, Yeo GS, Tschop MH, et al. POMC neuronal heterogeneity in energy balance and beyond: an integrated view. Nat Metab 2021;3:299–308.ArticlePubMedPMCPDF
- 4. Schneeberger M, Gomis R, Claret M. Hypothalamic and brainstem neuronal circuits controlling homeostatic energy balance. J Endocrinol 2014;220:T25–46.ArticlePubMed
- 5. Watts AG, Kanoski SE, Sanchez-Watts G, Langhans W. The physiological control of eating: signals, neurons, and networks. Physiol Rev 2022;102:689–813.ArticlePubMedPMC
- 6. Ruud J, Steculorum SM, Bruning JC. Neuronal control of peripheral insulin sensitivity and glucose metabolism. Nat Commun 2017;8:15259.ArticlePubMedPMCPDF
- 7. Pozo M, Claret M. Hypothalamic control of systemic glucose homeostasis: the pancreas connection. Trends Endocrinol Metab 2018;29:581–94.ArticlePubMed
- 8. Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol 2012;13:251–62.ArticlePubMedPMCPDF
- 9. Rutter GA, Da Silva Xavier G, Leclerc I. Roles of 5´-AMP-activated protein kinase (AMPK) in mammalian glucose homoeostasis. Biochem J 2003;375(Pt 1):1–16.ArticlePubMedPMCPDF
- 10. Srivastava RA, Pinkosky SL, Filippov S, Hanselman JC, Cramer CT, Newton RS. AMP-activated protein kinase: an emerging drug target to regulate imbalances in lipid and carbohydrate metabolism to treat cardio-metabolic diseases. J Lipid Res 2012;53:2490–514.ArticlePubMedPMC
- 11. Yang CS, Lam CK, Chari M, Cheung GW, Kokorovic A, Gao S, et al. Hypothalamic AMP-activated protein kinase regulates glucose production. Diabetes 2010;59:2435–43.ArticlePubMedPMCPDF
- 12. Han SM, Namkoong C, Jang PG, Park IS, Hong SW, Katakami H, et al. Hypothalamic AMP-activated protein kinase mediates counter-regulatory responses to hypoglycaemia in rats. Diabetologia 2005;48:2170–8.ArticlePubMedPDF
- 13. Hardie DG. AMPK: sensing energy while talking to other signaling pathways. Cell Metab 2014;20:939–52.ArticlePubMedPMC
- 14. Wang B, Cheng KK. Hypothalamic AMPK as a mediator of hormonal regulation of energy balance. Int J Mol Sci 2018;19:3552.ArticlePubMedPMC
- 15. Patel N, Khayat ZA, Ruderman NB, Klip A. Dissociation of 5´ AMP-activated protein kinase activation and glucose uptake stimulation by mitochondrial uncoupling and hyperosmolar stress: differential sensitivities to intracellular Ca2+ and protein kinase C inhibition. Biochem Biophys Res Commun 2001;285:1066–70.ArticlePubMed
- 16. Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest 2001;108:1167–74.ArticlePubMedPMC
- 17. Schneeberger M, Claret M. Recent insights into the role of hypothalamic AMPK signaling cascade upon metabolic control. Front Neurosci 2012;6:185.ArticlePubMedPMC
- 18. Xue B, Kahn BB. AMPK integrates nutrient and hormonal signals to regulate food intake and energy balance through effects in the hypothalamus and peripheral tissues. J Physiol 2006;574(Pt 1):73–83.ArticlePubMedPMC
- 19. Minokoshi Y, Alquier T, Furukawa N, Kim YB, Lee A, Xue B, et al. AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature 2004;428:569–74.ArticlePubMedPDF
- 20. Liu H, Xu Y, Hu F. AMPK in the ventromedial nucleus of the hypothalamus: a key regulator for thermogenesis. Front Endocrinol (Lausanne) 2020;11:578830.ArticlePubMedPMC
- 21. Lopez M, Tena-Sempere M. Estradiol effects on hypothalamic AMPK and BAT thermogenesis: a gateway for obesity treatment? Pharmacol Ther 2017;178:109–22.ArticlePubMed
- 22. Kim MS, Park JY, Namkoong C, Jang PG, Ryu JW, Song HS, et al. Anti-obesity effects of alpha-lipoic acid mediated by suppression of hypothalamic AMP-activated protein kinase. Nat Med 2004;10:727–33.ArticlePubMedPDF
- 23. Oh TS, Cho H, Cho JH, Yu SW, Kim EK. Hypothalamic AMPK-induced autophagy increases food intake by regulating NPY and POMC expression. Autophagy 2016;12:2009–25.ArticlePubMedPMC
- 24. Lopez M. Hypothalamic AMPK and energy balance. Eur J Clin Invest 2018;48:e12996.PubMedPMC
- 25. Kola B. Role of AMP-activated protein kinase in the control of appetite. J Neuroendocrinol 2008;20:942–51.ArticlePubMedPMC
- 26. Claret M, Smith MA, Batterham RL, Selman C, Choudhury AI, Fryer LG, et al. AMPK is essential for energy homeostasis regulation and glucose sensing by POMC and AgRP neurons. J Clin Invest 2007;117:2325–36.ArticlePubMedPMC
- 27. Martinez de Morentin PB, Gonzalez-Garcia I, Martins L, Lage R, Fernandez-Mallo D, Martinez-Sanchez N, et al. Estradiol regulates brown adipose tissue thermogenesis via hypothalamic AMPK. Cell Metab 2014;20:41–53.ArticlePubMedPMC
- 28. Martinez-Sanchez N, Seoane-Collazo P, Contreras C, Varela L, Villarroya J, Rial-Pensado E, et al. Hypothalamic AMPK-ER stress-JNK1 axis mediates the central actions of thyroid hormones on energy balance. Cell Metab 2017;26:212–29.ArticlePubMedPMC
- 29. Lam CK, Chari M, Rutter GA, Lam TK. Hypothalamic nutrient sensing activates a forebrain-hindbrain neuronal circuit to regulate glucose production in vivo. Diabetes 2011;60:107–13.ArticlePubMedPMCPDF
- 30. Perrin C, Knauf C, Burcelin R. Intracerebroventricular infusion of glucose, insulin, and the adenosine monophosphate-activated kinase activator, 5-aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside, controls muscle glycogen synthesis. Endocrinology 2004;145:4025–33.ArticlePubMed
- 31. Viollet B, Andreelli F, Jorgensen SB, Perrin C, Geloen A, Flamez D, et al. The AMP-activated protein kinase alpha2 catalytic subunit controls whole-body insulin sensitivity. J Clin Invest 2003;111:91–8.ArticlePubMedPMC
- 32. Friedman JM, Halaas JL. Leptin and the regulation of body weight in mammals. Nature 1998;395:763–70.ArticlePubMedPDF
- 33. Cheung CC, Clifton DK, Steiner RA. Proopiomelanocortin neurons are direct targets for leptin in the hypothalamus. Endocrinology 1997;138:4489–92.ArticlePubMed
- 34. Hakansson ML, Brown H, Ghilardi N, Skoda RC, Meister B. Leptin receptor immunoreactivity in chemically defined target neurons of the hypothalamus. J Neurosci 1998;18:559–72.ArticlePubMedPMC
- 35. Mercer JG, Hoggard N, Williams LM, Lawrence CB, Hannah LT, Morgan PJ, et al. Coexpression of leptin receptor and preproneuropeptide Y mRNA in arcuate nucleus of mouse hypothalamus. J Neuroendocrinol 1996;8:733–5.ArticlePubMed
- 36. Pelleymounter MA, Cullen MJ, Baker MB, Hecht R, Winters D, Boone T, et al. Effects of the obese gene product on body weight regulation in ob/ob mice. Science 1995;269:540–3.ArticlePubMed
- 37. Chen H, Charlat O, Tartaglia LA, Woolf EA, Weng X, Ellis SJ, et al. Evidence that the diabetes gene encodes the leptin receptor: identification of a mutation in the leptin receptor gene in db/db mice. Cell 1996;84:491–5.ArticlePubMed
- 38. Clement K, Vaisse C, Lahlou N, Cabrol S, Pelloux V, Cassuto D, et al. A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Nature 1998;392:398–401.ArticlePubMedPDF
- 39. Minokoshi Y, Kim YB, Peroni OD, Fryer LG, Muller C, Carling D, et al. Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature 2002;415:339–43.ArticlePubMedPDF
- 40. Obici S, Feng Z, Arduini A, Conti R, Rossetti L. Inhibition of hypothalamic carnitine palmitoyltransferase-1 decreases food intake and glucose production. Nat Med 2003;9:756–61.ArticlePubMedPDF
- 41. Gao S, Kinzig KP, Aja S, Scott KA, Keung W, Kelly S, et al. Leptin activates hypothalamic acetyl-CoA carboxylase to inhibit food intake. Proc Natl Acad Sci U S A 2007;104:17358–63.ArticlePubMedPMC
- 42. Sipols AJ, Baskin DG, Schwartz MW. Effect of intracerebroventricular insulin infusion on diabetic hyperphagia and hypothalamic neuropeptide gene expression. Diabetes 1995;44:147–51.ArticlePubMed
- 43. Schwartz MW, Figlewicz DP, Baskin DG, Woods SC, Porte D Jr. Insulin in the brain: a hormonal regulator of energy balance. Endocr Rev 1992;13:387–414.ArticlePubMed
- 44. Bruning JC, Gautam D, Burks DJ, Gillette J, Schubert M, Orban PC, et al. Role of brain insulin receptor in control of body weight and reproduction. Science 2000;289:2122–5.ArticlePubMed
- 45. Woods SC, Lotter EC, McKay LD, Porte D Jr. Chronic intracerebroventricular infusion of insulin reduces food intake and body weight of baboons. Nature 1979;282:503–5.ArticlePubMedPDF
- 46. Woods SC, Seeley RJ, Porte D Jr, Schwartz MW. Signals that regulate food intake and energy homeostasis. Science 1998;280:1378–83.ArticlePubMed
- 47. Richardson RD, Ramsay DS, Lernmark A, Scheurink AJ, Baskin DG, Woods SC. Weight loss in rats following intraventricular transplants of pancreatic islets. Am J Physiol 1994;266(1 Pt 2):R59–64.ArticlePubMed
- 48. Air EL, Strowski MZ, Benoit SC, Conarello SL, Salituro GM, Guan XM, et al. Small molecule insulin mimetics reduce food intake and body weight and prevent development of obesity. Nat Med 2002;8:179–83.ArticlePubMedPDF
- 49. Namkoong C, Kim MS, Jang PG, Han SM, Park HS, Koh EH, et al. Enhanced hypothalamic AMP-activated protein kinase activity contributes to hyperphagia in diabetic rats. Diabetes 2005;54:63–8.ArticlePubMedPDF
- 50. Quaresma PG, Reencober N, Zanotto TM, Santos AC, Weissmann L, de Matos AH, et al. Pioglitazone treatment increases food intake and decreases energy expenditure partially via hypothalamic adiponectin/adipoR1/AMPK pathway. Int J Obes (Lond) 2016;40:138–46.ArticlePubMedPDF
- 51. Wren AM, Seal LJ, Cohen MA, Brynes AE, Frost GS, Murphy KG, et al. Ghrelin enhances appetite and increases food intake in humans. J Clin Endocrinol Metab 2001;86:5992.ArticlePubMed
- 52. Chen HY, Trumbauer ME, Chen AS, Weingarth DT, Adams JR, Frazier EG, et al. Orexigenic action of peripheral ghrelin is mediated by neuropeptide Y and agouti-related protein. Endocrinology 2004;145:2607–12.ArticlePubMed
- 53. Andersson U, Filipsson K, Abbott CR, Woods A, Smith K, Bloom SR, et al. AMP-activated protein kinase plays a role in the control of food intake. J Biol Chem 2004;279:12005–8.ArticlePubMed
- 54. Lopez M, Lage R, Saha AK, Perez-Tilve D, Vazquez MJ, Varela L, et al. Hypothalamic fatty acid metabolism mediates the orexigenic action of ghrelin. Cell Metab 2008;7:389–99.ArticlePubMed
- 55. Gao S, Casals N, Keung W, Moran TH, Lopaschuk GD. Differential effects of central ghrelin on fatty acid metabolism in hypothalamic ventral medial and arcuate nuclei. Physiol Behav 2013;118:165–70.ArticlePubMed
- 56. Lim CT, Kola B, Feltrin D, Perez-Tilve D, Tschop MH, Grossman AB, et al. Ghrelin and cannabinoids require the ghrelin receptor to affect cellular energy metabolism. Mol Cell Endocrinol 2013;365:303–8.ArticlePubMedPMC
- 57. Andrews ZB, Liu ZW, Walllingford N, Erion DM, Borok E, Friedman JM, et al. UCP2 mediates ghrelin’s action on NPY/AgRP neurons by lowering free radicals. Nature 2008;454:846–51.ArticlePubMedPMCPDF
- 58. Kola B, Hubina E, Tucci SA, Kirkham TC, Garcia EA, Mitchell SE, et al. Cannabinoids and ghrelin have both central and peripheral metabolic and cardiac effects via AMP-activated protein kinase. J Biol Chem 2005;280:25196–201.ArticlePubMed
- 59. Wren AM, Small CJ, Ward HL, Murphy KG, Dakin CL, Taheri S, et al. The novel hypothalamic peptide ghrelin stimulates food intake and growth hormone secretion. Endocrinology 2000;141:4325–8.ArticlePubMed
- 60. Yang Y, Atasoy D, Su HH, Sternson SM. Hunger states switch a flip-flop memory circuit via a synaptic AMPK-dependent positive feedback loop. Cell 2011;146:992–1003.ArticlePubMedPMC
- 61. Andrews ZB. Central mechanisms involved in the orexigenic actions of ghrelin. Peptides 2011;32:2248–55.ArticlePubMed
- 62. Kohno D, Sone H, Minokoshi Y, Yada T. Ghrelin raises [Ca2+]i via AMPK in hypothalamic arcuate nucleus NPY neurons. Biochem Biophys Res Commun 2008;366:388–92.ArticlePubMed
- 63. Goldstone AP, Morgan I, Mercer JG, Morgan DG, Moar KM, Ghatei MA, et al. Effect of leptin on hypothalamic GLP-1 peptide and brain-stem pre-proglucagon mRNA. Biochem Biophys Res Commun 2000;269:331–5.ArticlePubMed
- 64. Trapp S, Richards JE. The gut hormone glucagon-like peptide-1 produced in brain: is this physiologically relevant? Curr Opin Pharmacol 2013;13:964–9.ArticlePubMedPMC
- 65. Turton MD, O’Shea D, Gunn I, Beak SA, Edwards CM, Meeran K, et al. A role for glucagon-like peptide-1 in the central regulation of feeding. Nature 1996;379:69–72.ArticlePubMedPDF
- 66. Seo S, Ju S, Chung H, Lee D, Park S. Acute effects of glucagon-like peptide-1 on hypothalamic neuropeptide and AMP activated kinase expression in fasted rats. Endocr J 2008;55:867–74.ArticlePubMed
- 67. Hurtado-Carneiro V, Sanz C, Roncero I, Vazquez P, Blazquez E, Alvarez E. Glucagon-like peptide 1 (GLP-1) can reverse AMP-activated protein kinase (AMPK) and S6 kinase (P70S6K) activities induced by fluctuations in glucose levels in hypothalamic areas involved in feeding behaviour. Mol Neurobiol 2012;45:348–61.ArticlePubMedPDF
- 68. Beiroa D, Imbernon M, Gallego R, Senra A, Herranz D, Villarroya F, et al. GLP-1 agonism stimulates brown adipose tissue thermogenesis and browning through hypothalamic AMPK. Diabetes 2014;63:3346–58.ArticlePubMedPDF
- 69. Burmeister MA, Brown JD, Ayala JE, Stoffers DA, Sandoval DA, Seeley RJ, et al. The glucagon-like peptide-1 receptor in the ventromedial hypothalamus reduces short-term food intake in male mice by regulating nutrient sensor activity. Am J Physiol Endocrinol Metab 2017;313:E651–62.ArticlePubMedPMC
- 70. Wang ZV, Scherer PE. Adiponectin, the past two decades. J Mol Cell Biol 2016;8:93–100.ArticlePubMedPMC
- 71. Cheng KK, Lam KS, Wang B, Xu A. Signaling mechanisms underlying the insulin-sensitizing effects of adiponectin. Best Pract Res Clin Endocrinol Metab 2014;28:3–13.ArticlePubMed
- 72. Hotta K, Funahashi T, Arita Y, Takahashi M, Matsuda M, Okamoto Y, et al. Plasma concentrations of a novel, adipose-specific protein, adiponectin, in type 2 diabetic patients. Arterioscler Thromb Vasc Biol 2000;20:1595–9.ArticlePubMed
- 73. Weyer C, Funahashi T, Tanaka S, Hotta K, Matsuzawa Y, Pratley RE, et al. Hypoadiponectinemia in obesity and type 2 diabetes: close association with insulin resistance and hyperinsulinemia. J Clin Endocrinol Metab 2001;86:1930–5.ArticlePubMed
- 74. Thundyil J, Pavlovski D, Sobey CG, Arumugam TV. Adiponectin receptor signalling in the brain. Br J Pharmacol 2012;165:313–27.ArticlePubMedPMC
- 75. Kubota N, Yano W, Kubota T, Yamauchi T, Itoh S, Kumagai H, et al. Adiponectin stimulates AMP-activated protein kinase in the hypothalamus and increases food intake. Cell Metab 2007;6:55–68.PubMed
- 76. Steinberg GR, Carling D. AMP-activated protein kinase: the current landscape for drug development. Nat Rev Drug Discov 2019;18:527–51.ArticlePubMedPDF
- 77. Watt MJ, Holmes AG, Pinnamaneni SK, Garnham AP, Steinberg GR, Kemp BE, et al. Regulation of HSL serine phosphorylation in skeletal muscle and adipose tissue. Am J Physiol Endocrinol Metab 2006;290:E500–8.ArticlePubMed
- 78. Ahmadian M, Abbott MJ, Tang T, Hudak CS, Kim Y, Bruss M, et al. Desnutrin/ATGL is regulated by AMPK and is required for a brown adipose phenotype. Cell Metab 2011;13:739–48.ArticlePubMedPMC
- 79. Knudsen JR, Persson KW, Henriquez-Olguin C, Li Z, Di Leo N, Hesselager SA, et al. Microtubule-mediated GLUT4 trafficking is disrupted in insulin-resistant skeletal muscle. Elife 2023;12:e83338.PubMedPMC
- 80. El-Mir MY, Nogueira V, Fontaine E, Averet N, Rigoulet M, Leverve X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J Biol Chem 2000;275:223–8.ArticlePubMed
- 81. Gan SC, Barr J, Arieff AI, Pearl RG. Biguanide-associated lactic acidosis: case report and review of the literature. Arch Intern Med 1992;152:2333–6.ArticlePubMed
- 82. Chan NN, Brain HP, Feher MD. Metformin-associated lactic acidosis: a rare or very rare clinical entity? Diabet Med 1999;16:273–81.ArticlePubMedPDF
- 83. Duan Y, Zhang R, Zhang M, Sun L, Dong S, Wang G, et al. Metformin inhibits food intake and neuropeptide Y gene expression in the hypothalamus. Neural Regen Res 2013;8:2379–88.PubMedPMC
- 84. Stevanovic D, Janjetovic K, Misirkic M, Vucicevic L, Sumarac-Dumanovic M, Micic D, et al. Intracerebroventricular administration of metformin inhibits ghrelin-induced hypothalamic AMP-kinase signalling and food intake. Neuroendocrinology 2012;96:24–31.ArticlePubMedPDF
- 85. Fryer LG, Parbu-Patel A, Carling D. The anti-diabetic drugs rosiglitazone and metformin stimulate AMP-activated protein kinase through distinct signaling pathways. J Biol Chem 2002;277:25226–32.ArticlePubMed
- 86. Saha AK, Avilucea PR, Ye JM, Assifi MM, Kraegen EW, Ruderman NB. Pioglitazone treatment activates AMP-activated protein kinase in rat liver and adipose tissue in vivo. Biochem Biophys Res Commun 2004;314:580–5.ArticlePubMed
- 87. Brunmair B, Staniek K, Gras F, Scharf N, Althaym A, Clara R, et al. Thiazolidinediones, like metformin, inhibit respiratory complex I: a common mechanism contributing to their antidiabetic actions? Diabetes 2004;53:1052–9.PubMed
- 88. Lee WJ, Song KH, Koh EH, Won JC, Kim HS, Park HS, et al. Alpha-lipoic acid increases insulin sensitivity by activating AMPK in skeletal muscle. Biochem Biophys Res Commun 2005;332:885–91.PubMed
- 89. Lee Y, Naseem RH, Park BH, Garry DJ, Richardson JA, Schaffer JE, et al. Alpha-lipoic acid prevents lipotoxic cardiomyopathy in acyl CoA-synthase transgenic mice. Biochem Biophys Res Commun 2006;344:446–52.PubMed
- 90. Lee WJ, Lee IK, Kim HS, Kim YM, Koh EH, Won JC, et al. Alpha-lipoic acid prevents endothelial dysfunction in obese rats via activation of AMP-activated protein kinase. Arterioscler Thromb Vasc Biol 2005;25:2488–94.PubMed
- 91. Shen QW, Zhu MJ, Tong J, Ren J, Du M. Ca2+/calmodulin-dependent protein kinase kinase is involved in AMP-activated protein kinase activation by alpha-lipoic acid in C2C12 myotubes. Am J Physiol Cell Physiol 2007;293:C1395–403.PubMed
- 92. Hardie DG. AMP-activated protein kinase as a drug target. Annu Rev Pharmacol Toxicol 2007;47:185–210.ArticlePubMed
- 93. Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol 2011;13:1016–23.ArticlePubMedPMCPDF
- 94. Kim J, Yang G, Kim Y, Kim J, Ha J. AMPK activators: mechanisms of action and physiological activities. Exp Mol Med 2016;48:e224.ArticlePubMedPMCPDF
- 95. Tukhovskaya EA, Shaykhutdinova ER, Pakhomova IA, Slashcheva GA, Goryacheva NA, Sadovnikova ES, et al. AICAR improves outcomes of metabolic syndrome and type 2 diabetes induced by high-fat diet in C57Bl/6 male mice. Int J Mol Sci 2022;23:15719.ArticlePubMedPMC
- 96. Boon H, Bosselaar M, Praet SF, Blaak EE, Saris WH, Wagenmakers AJ, et al. Intravenous AICAR administration reduces hepatic glucose output and inhibits whole body lipolysis in type 2 diabetic patients. Diabetologia 2008;51:1893–900.ArticlePubMed
- 97. de Jonge R, Macleod DC, Suryapranata H, van Es GA, Friedman J, Serruys PW, et al. Effect of acadesine on myocardial ischaemia in patients with coronary artery disease. Eur J Pharmacol 1997;337:41–4.ArticlePubMed
- 98. Newman MF, Ferguson TB, White JA, Ambrosio G, Koglin J, Nussmeier NA, et al. Effect of adenosine-regulating agent acadesine on morbidity and mortality associated with coronary artery bypass grafting: the RED-CABG randomized controlled trial. JAMA 2012;308:157–64.PubMed
- 99. Cluzeau T, Furstoss N, Savy C, El Manaa W, Zerhouni M, Blot L, et al. Acadesine circumvents azacitidine resistance in myelodysplastic syndrome and acute myeloid leukemia. Int J Mol Sci 2019;21:164.ArticlePubMedPMC
- 100. Cool B, Zinker B, Chiou W, Kifle L, Cao N, Perham M, et al. Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell Metab 2006;3:403–16.PubMed
- 101. Goransson O, McBride A, Hawley SA, Ross FA, Shpiro N, Foretz M, et al. Mechanism of action of A-769662, a valuable tool for activation of AMP-activated protein kinase. J Biol Chem 2007;282:32549–60.ArticlePubMedPMC
- 102. Pinkosky SL, Scott JW, Desjardins EM, Smith BK, Day EA, Ford RJ, et al. Long-chain fatty acyl-CoA esters regulate metabolism via allosteric control of AMPK β1 isoforms. Nat Metab 2020;2:873–81.ArticlePubMedPMCPDF
- 103. Saito M, Saito M, Das BC. Involvement of AMP-activated protein kinase in neuroinflammation and neurodegeneration in the adult and developing brain. Int J Dev Neurosci 2019;77:48–59.ArticlePubMedPMCPDF
- 104. Xu Y, Xu Y, Wang Y, Wang Y, He L, Jiang Z, et al. Telmisartan prevention of LPS-induced microglia activation involves M2 microglia polarization via CaMKKβ-dependent AMPK activation. Brain Behav Immun 2015;50:298–313.ArticlePubMed
- 105. Barialai L, Strecker MI, Luger AL, Jager M, Bruns I, Sittig ACM, et al. AMPK activation protects astrocytes from hypoxia-induced cell death. Int J Mol Med 2020;45:1385–96.ArticlePubMedPMC
- 106. Muraleedharan R, Gawali MV, Tiwari D, Sukumaran A, Oatman N, Anderson J, et al. AMPK-regulated astrocytic lactate shuttle plays a non-cell-autonomous role in neuronal survival. Cell Rep 2020;32:108092.ArticlePubMedPMC
 PubReader
PubReader ePub Link
ePub Link Cite
Cite