
Search
- Page Path
- HOME > Search
Original Articles
- Adrenal Gland
Big Data Articles (National Health Insurance Service Database) - Epidemiology and Long-Term Adverse Outcomes in Korean Patients with Congenital Adrenal Hyperplasia: A Nationwide Study
- Jung Hee Kim, Sunkyu Choi, Young Ah Lee, Juneyoung Lee, Sin Gon Kim
- Endocrinol Metab. 2022;37(1):138-147. Published online February 28, 2022
- DOI: https://doi.org/10.3803/EnM.2021.1328
- 3,382 View
- 149 Download
- 8 Web of Science
- 10 Crossref
-
 Abstract
Abstract
 PDF
PDF PubReader
PubReader  ePub
ePub - Background
Previous studies on the epidemiology and complications of congenital adrenal hyperplasia (CAH) were conducted in Western countries and in children/adolescents. We aimed to explore the epidemiology of CAH, as well as the risk of comorbidities and mortality, in a Korean nationwide case-control study.
Methods
CAH patients (n=2,840) were included between 2002 and 2017 from the National Health Insurance Service database and the Rare Intractable Disease program. CAH patients were compared, at a 1:10 ratio, with age-, sex-, and index year-matched controls (n=28,400).
Results
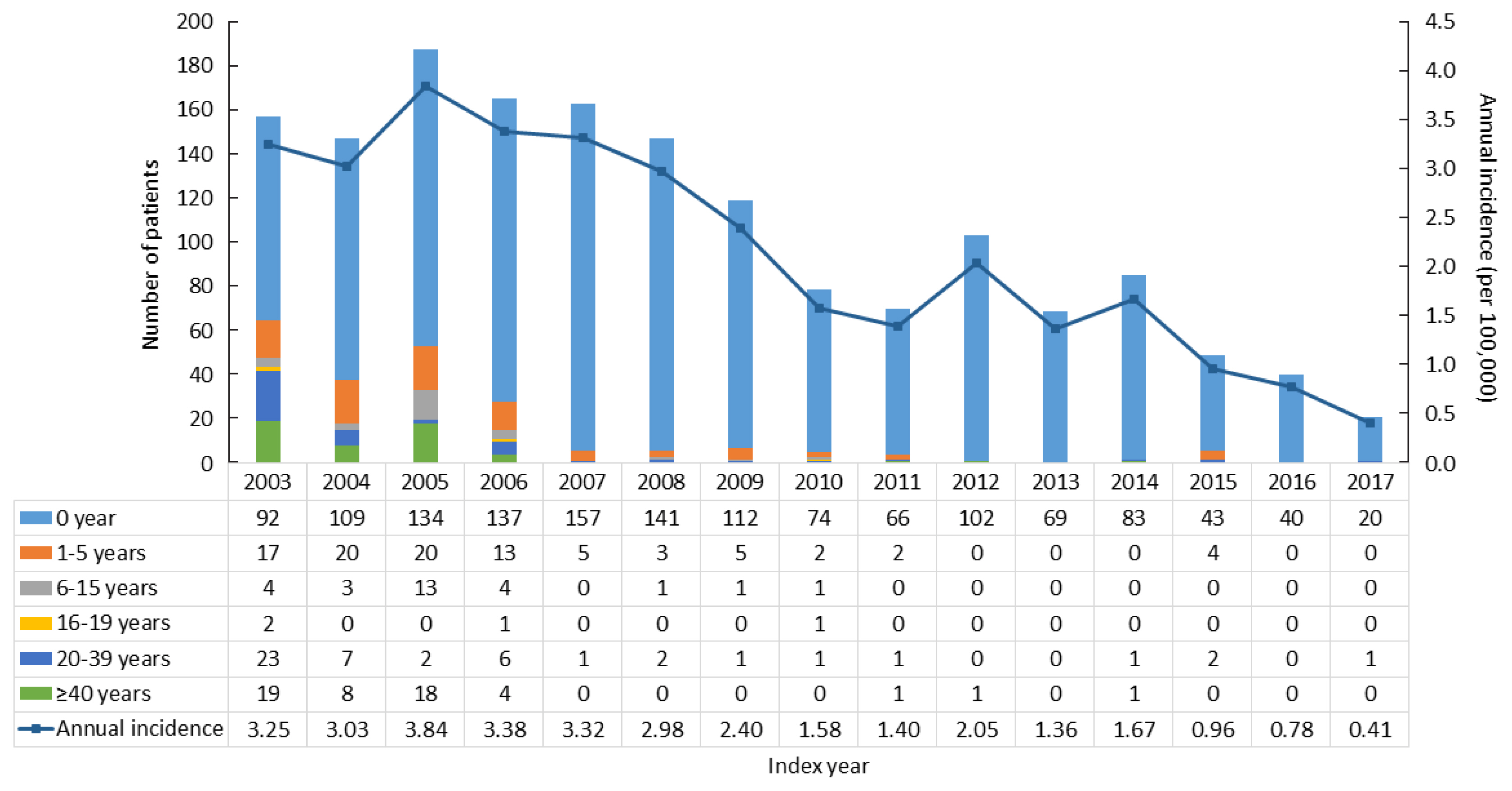
The point prevalence of CAH patients in Korea was 1 in 18,745 persons in 2017. The annual incidence rate declined between 2003 and 2017 from 3.25 to 0.41 per 100,000 persons. CAH patients were at elevated risk for cardiovascular disease (odds ratio [OR], 1.6; 95% confidence interval [CI], 1.4 to 1.9), stroke (OR, 1.7; 95% CI, 1.3 to 2.0), diabetes mellitus (OR, 2.8; 95% CI, 2.6 to 3.1), dyslipidemia (OR, 2.4; 95% CI, 2.2 to 2.6), and psychiatric disorders (OR, 1.5; 95% CI, 1.3 to 1.6). Fracture risk increased in CAH patients aged over 40 years (OR, 1.4; 95% CI, 1.1 to 1.7). CAH patients were at higher risk of mortality than controls (hazard ratio, 1.6; 95% CI, 1.3 to 2.0).
Conclusion
Our nationwide study showed a recent decline in the incidence of CAH and an elevated risk for cardiovascular, metabolic, skeletal, and psychiatric disorders in CAH patients. Lifelong management for comorbidity risk is a crucial component of treating CAH patients. -
Citations
Citations to this article as recorded by
- Hyperandrogenism and Cardiometabolic Risk in Pre- and Postmenopausal Women—What Is the Evidence?
Angelica Lindén Hirschberg
The Journal of Clinical Endocrinology & Metabolism.2024; 109(5): 1202. CrossRef - Predictors of Cardiovascular Morbidities in Adults With 21-Hydroxylase Deficiency Congenital Adrenal Hyperplasia
Suranut Charoensri, Richard J Auchus
The Journal of Clinical Endocrinology & Metabolism.2024; 109(3): e1133. CrossRef - Case report: Development of central precocious puberty in a girl with late-diagnosed simple virilizing congenital adrenal hyperplasia complicated with Williams syndrome
Eun Young Joo, Myung Ji Yoo, Su Jin Kim, Woori Jang, Ji-Eun Lee
Frontiers in Endocrinology.2024;[Epub] CrossRef - Анализ распространенности и заболеваемости надпочечниковой недостаточностью в мире
М. Ю. Юкина, Н. Ф. Нуралиева, Е. А. Трошина
Ateroscleroz.2023; 18(4): 426. CrossRef - Big Data Research in the Field of Endocrine Diseases Using the Korean National Health Information Database
Sun Wook Cho, Jung Hee Kim, Han Seok Choi, Hwa Young Ahn, Mee Kyoung Kim, Eun Jung Rhee
Endocrinology and Metabolism.2023; 38(1): 10. CrossRef - Long-term cardiometabolic morbidity in young adults with classic 21-hydroxylase deficiency congenital adrenal hyperplasia
Beatrice Righi, Salma R. Ali, Jillian Bryce, Jeremy W. Tomlinson, Walter Bonfig, Federico Baronio, Eduardo C. Costa, Guilherme Guaragna-Filho, Guy T’Sjoen, Martine Cools, Renata Markosyan, Tania A. S. S. Bachega, Mirela C. Miranda, Violeta Iotova, Henrik
Endocrine.2023; 80(3): 630. CrossRef - Serum steroid profile captures metabolic phenotypes in adults with classic congenital adrenal hyperplasia
Chang Ho Ahn, Jaeyoon Shim, Han Na Jang, Young Ah Lee, Sang-Won Lee, Man Ho Choi, Jung Hee Kim
The Journal of Steroid Biochemistry and Molecular Biology.2023; 234: 106374. CrossRef - Long‐term health consequences of congenital adrenal hyperplasia
Riccardo Pofi, Xiaochen Ji, Nils P. Krone, Jeremy W. Tomlinson
Clinical Endocrinology.2023;[Epub] CrossRef - Multiplexed Serum Steroid Profiling Reveals Metabolic Signatures of Subtypes in Congenital Adrenal Hyperplasia
Jaeyoon Shim, Chang Ho Ahn, Seung Shin Park, Jongsung Noh, Chaelin Lee, Sang Won Lee, Jung Hee Kim, Man Ho Choi
Journal of the Endocrine Society.2023;[Epub] CrossRef - Long-Term Outcomes of Congenital Adrenal Hyperplasia
Anna Nordenström, Svetlana Lajic, Henrik Falhammar
Endocrinology and Metabolism.2022; 37(4): 587. CrossRef
- Hyperandrogenism and Cardiometabolic Risk in Pre- and Postmenopausal Women—What Is the Evidence?

- Adrenal Gland
- Adrenal Morphology as an Indicator of Long-Term Disease Control in Adults with Classic 21-Hydroxylase Deficiency
- Taek Min Kim, Jung Hee Kim, Han Na Jang, Man Ho Choi, Jeong Yeon Cho, Sang Youn Kim
- Endocrinol Metab. 2022;37(1):124-137. Published online February 8, 2022
- DOI: https://doi.org/10.3803/EnM.2021.1278
- 4,271 View
- 126 Download
- 6 Web of Science
- 6 Crossref
-
Abstract
PDFPubReader ePub
- Background
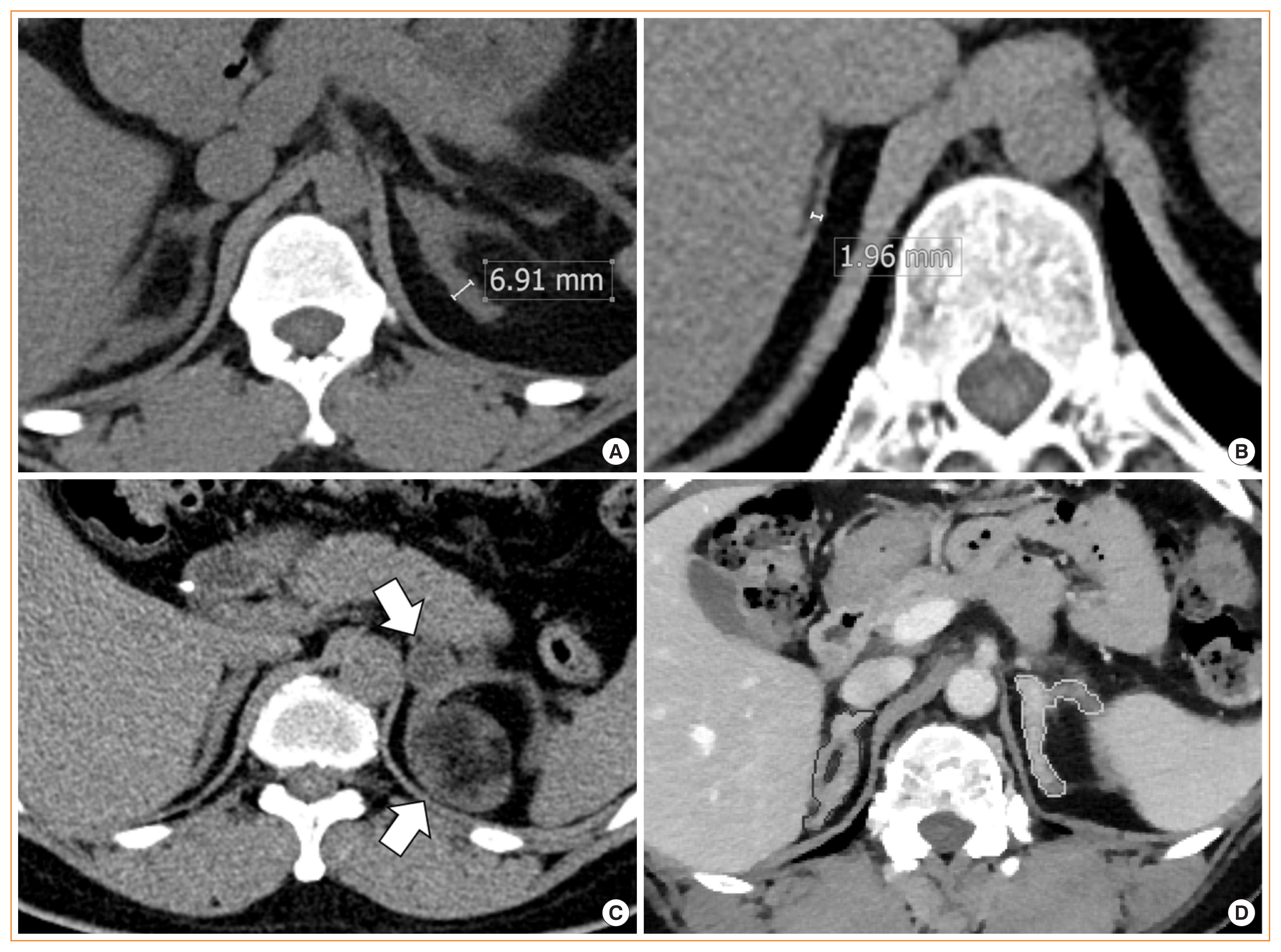
Monitoring adults with classical 21-hydroxylase deficiency (21OHD) is challenging due to variation in clinical and laboratory settings. Moreover, guidelines for adrenal imaging in 21OHD are not yet available. We evaluated the relationship between adrenal morphology and disease control status in classical 21OHD.
Methods
This retrospective, cross-sectional study included 90 adult 21OHD patients and 270 age- and sex-matched healthy controls. We assessed adrenal volume, width, and tumor presence using abdominal computed tomography and evaluated correlations of adrenal volume and width with hormonal status. We investigated the diagnostic performance of adrenal volume and width for identifying well-controlled status in 21OHD patients (17α-hydroxyprogesterone [17-OHP] <10 ng/mL).
Results
The adrenal morphology of 21OHD patients showed hypertrophy (45.6%), normal size (42.2%), and hypotrophy (12.2%). Adrenal tumors were detected in 12 patients (13.3%). The adrenal volume and width of 21OHD patients were significantly larger than those of controls (18.2±12.2 mL vs. 7.1±2.0 mL, 4.7±1.9 mm vs. 3.3±0.5 mm, P<0.001 for both). The 17-OHP and androstenedione levels were highest in patients with adrenal hypertrophy, followed by those with normal adrenal glands and adrenal hypotrophy (P<0.05 for both). Adrenal volume and width correlated positively with adrenocorticotropic hormone, 17-OHP, 11β-hydroxytestosterone, progesterone sulfate, and dehydroepiandrosterone sulfate in both sexes (r=0.33–0.95, P<0.05 for all). For identifying well-controlled patients, the optimal cut-off values of adrenal volume and width were 10.7 mL and 4 mm, respectively (area under the curve, 0.82–0.88; P<0.001 for both).
Conclusion
Adrenal volume and width may be reliable quantitative parameters for monitoring patients with classical 21OHD. -
Citations
Citations to this article as recorded by- Long‐term health consequences of congenital adrenal hyperplasia
Riccardo Pofi, Xiaochen Ji, Nils P. Krone, Jeremy W. Tomlinson
Clinical Endocrinology.2023;[Epub] CrossRef - Landscape of Adrenal Tumours in Patients with Congenital Adrenal Hyperplasia
Mara Carsote, Ana-Maria Gheorghe, Claudiu Nistor, Alexandra-Ioana Trandafir, Oana-Claudia Sima, Anca-Pati Cucu, Adrian Ciuche, Eugenia Petrova, Adina Ghemigian
Biomedicines.2023; 11(11): 3081. CrossRef - Multiplexed Serum Steroid Profiling Reveals Metabolic Signatures of Subtypes in Congenital Adrenal Hyperplasia
Jaeyoon Shim, Chang Ho Ahn, Seung Shin Park, Jongsung Noh, Chaelin Lee, Sang Won Lee, Jung Hee Kim, Man Ho Choi
Journal of the Endocrine Society.2023;[Epub] CrossRef - Long-Term Outcomes of Congenital Adrenal Hyperplasia
Anna Nordenström, Svetlana Lajic, Henrik Falhammar
Endocrinology and Metabolism.2022; 37(4): 587. CrossRef - Congenital adrenal hyperplasia in patients with adrenal tumors: a population-based case–control study
F. Sahlander, J. Patrova, B. Mannheimer, J. D. Lindh, H. Falhammar
Journal of Endocrinological Investigation.2022; 46(3): 559. CrossRef - Fully automatic volume measurement of the adrenal gland on CT using deep learning to classify adrenal hyperplasia
Taek Min Kim, Seung Jae Choi, Ji Yeon Ko, Sungwan Kim, Chang Wook Jeong, Jeong Yeon Cho, Sang Youn Kim, Young-Gon Kim
European Radiology.2022; 33(6): 4292. CrossRef
- Long‐term health consequences of congenital adrenal hyperplasia
Review Articles
- Thyroid
- Antithyroid Drug Treatment in Graves’ Disease
- Jae Hoon Chung
- Endocrinol Metab. 2021;36(3):491-499. Published online June 16, 2021
- DOI: https://doi.org/10.3803/EnM.2021.1070
- 5,028 View
- 338 Download
- 6 Web of Science
- 9 Crossref
-
Abstract
PDFPubReader ePub
- Graves’ disease is associated with thyrotropin (TSH) receptor stimulating antibody, for which there is no therapeutic agent. This disease is currently treated through inhibition of thyroid hormone synthesis or destruction of the thyroid gland. Recurrence after antithyroid drug (ATD) treatment is common. Recent studies have shown that the longer is the duration of use of ATD, the higher is the remission rate. Considering the relationship between clinical outcomes and iodine intake, recurrence of Graves’ disease is more common in iodine-deficient areas than in iodine-sufficient areas. Iodine restriction in an iodine-excessive area does not improve the effectiveness of ATD or increase remission rates. Recently, Danish and Korean nationwide studies noted significantly higher prevalence of birth defects in newborns exposed to ATD during the first trimester compared to that of those who did not have such exposure. The prevalence of birth defects was lowest when propylthiouracil (PTU) was used and decreased by only 0.15% when methimazole was changed to PTU in the first trimester. Therefore, it is best not to use ATD in the first trimester or to change to PTU before pregnancy.
-
Citations
Citations to this article as recorded by- Выраженность окислительного стресса и энзиматическая активность нейтрофилов крови у пациентов с болезнью Грейвса в зависимости от компенсации гипертиреоза
М. А. Дудина, С. А. Догадин, А. А. Савченко, И. И. Гвоздев
Ateroscleroz.2023; 18(4): 411. CrossRef - Application of oral inorganic iodine in the treatment of Graves’ disease
Yixuan Huang, Yihang Xu, Murong Xu, Xiaotong Zhao, Mingwei Chen
Frontiers in Endocrinology.2023;[Epub] CrossRef - Sex-specific risk factors associated with graves’ orbitopathy in Korean patients with newly diagnosed graves’ disease
Jooyoung Lee, Jinmo Kang, Hwa Young Ahn, Jeong Kyu Lee
Eye.2023; 37(16): 3382. CrossRef - Methimazole, an Effective Neutralizing Agent of the Sulfur Mustard Derivative 2-Chloroethyl Ethyl Sulfide
Albert Armoo, Tanner Diemer, Abigail Donkor, Jerrod Fedorchik, Severine Van slambrouck, Rachel Willand-Charnley, Brian A. Logue
ACS Bio & Med Chem Au.2023; 3(5): 448. CrossRef - Increased risk of incident gout in patients with hyperthyroidism: a nationwide retrospective cohort study
Ju-Yeun Lee, So-Yeon Park, Seo Young Sohn
Rheumatology International.2023; 44(3): 451. CrossRef - The influence of thionamides on intra-thyroidal uptake of 131I during radioiodine-131 treatment of Graves’ disease
Christian Happel, Benjamin Bockisch, Britta Leonhäuser, Amir Sabet, Frank Grünwald, Daniel Groener
Scientific Reports.2023;[Epub] CrossRef - Usefulness of Real-Time Quantitative Microvascular Ultrasonography for Differentiation of Graves’ Disease from Destructive Thyroiditis in Thyrotoxic Patients
Han-Sang Baek, Ji-Yeon Park, Chai-Ho Jeong, Jeonghoon Ha, Moo Il Kang, Dong-Jun Lim
Endocrinology and Metabolism.2022; 37(2): 323. CrossRef - The chemiluminescent and enzymatic activity of blood neutrophils in patients with Graves' disease depending on hyperthyroidism compensation
M. A. Dudina, A. A. Savchenko, S. A. Dogadin, I. I. Gvozdev
Clinical and experimental thyroidology.2022; 18(1): 4. CrossRef - Risk of Diabetes in Patients with Long-Standing Graves’ Disease: A Longitudinal Study
Eyun Song, Min Ji Koo, Eunjin Noh, Soon Young Hwang, Min Jeong Park, Jung A Kim, Eun Roh, Kyung Mook Choi, Sei Hyun Baik, Geum Joon Cho, Hye Jin Yoo
Endocrinology and Metabolism.2021; 36(6): 1277. CrossRef
- Выраженность окислительного стресса и энзиматическая активность нейтрофилов крови у пациентов с болезнью Грейвса в зависимости от компенсации гипертиреоза
- Miscellaneous
- WD40-Repeat Proteins in Ciliopathies and Congenital Disorders of Endocrine System
- Yeonjoo Kim, Soo-Hyun Kim
- Endocrinol Metab. 2020;35(3):494-506. Published online September 8, 2020
- DOI: https://doi.org/10.3803/EnM.2020.302

- 14,875 View
- 199 Download
- 9 Web of Science
- 9 Crossref
-
Abstract
PDF
 Supplementary MaterialPubReader ePub
Supplementary MaterialPubReader ePub - WD40-repeat (WDR)-containing proteins constitute an evolutionarily conserved large protein family with a broad range of biological functions. In human proteome, WDR makes up one of the most abundant protein-protein interaction domains. Members of the WDR protein family play important roles in nearly all major cellular signalling pathways. Mutations of WDR proteins have been associated with various human pathologies including neurological disorders, cancer, obesity, ciliopathies and endocrine disorders. This review provides an updated overview of the biological functions of WDR proteins and their mutations found in congenital disorders. We also highlight the significant role of WDR proteins in ciliopathies and endocrine disorders. The new insights may help develop therapeutic approaches targeting WDR motifs.
-
Citations
Citations to this article as recorded by- Exosomes Derived from Bone Marrow Mesenchymal Stem Cells Alleviate Rheumatoid Arthritis Symptoms via Shuttling Proteins
Lijun Wang, Fei Li, Liting Wang, Bingxing Wu, Min Du, Hua Xing, Shifeng Pan
Journal of Proteome Research.2024; 23(4): 1298. CrossRef - Structural screens identify candidate human homologs of insect chemoreceptors and cryptic Drosophila gustatory receptor-like proteins
Richard Benton, Nathaniel J Himmel
eLife.2023;[Epub] CrossRef - Changes in protein phosphorylation by insulin administration in the central nervous system of the gastropod mollusk Lymnaea stagnalis
Junko Nakai, Kengo Namiki, Yuki Totani, Shigeki Yasumasu, Teruki Yoshimura, Takashi Aoki, Etsuro Ito
Biophysics and Physicobiology.2023; 20(4): n/a. CrossRef - Unveiling Distinct Proteomic Signatures in Complicated Crohn’s Disease That Could Predict the Disease Course
Laura A. Lucaciu, Radu Seicean, Alina Uifălean, Maria Iacobescu, Cristina A. Iuga, Andrada Seicean
International Journal of Molecular Sciences.2023; 24(23): 16966. CrossRef - Aromatic patterns: Tryptophan aromaticity as a catalyst for the emergence of life and rise of consciousness
Amal Alachkar
Physics of Life Reviews.2022; 42: 93. CrossRef - EML2-S constitutes a new class of proteins that recognizes and regulates the dynamics of tyrosinated microtubules
Takashi Hotta, Thomas S. McAlear, Yang Yue, Takumi Higaki, Sarah E. Haynes, Alexey I. Nesvizhskii, David Sept, Kristen J. Verhey, Susanne Bechstedt, Ryoma Ohi
Current Biology.2022; 32(18): 3898. CrossRef - Susceptibility of craniofacial ciliopathies to oral cancer-A proposed research
G Arun Kumar
Journal of Dental Health, Oral Disorders & Therapy.2022; 13(2): 41. CrossRef - A WDR47 homolog facilitates ciliogenesis by modulating intraflagellar transport
Chun-Xue Song, Xian-Ting Zeng, Wan-Xin Zeng, Rong Liu, Xia-Jing Tong, Qian Li
Journal of Cell Science.2022;[Epub] CrossRef - Biallelic loss-of-function variants in WDR11 are associated with microcephaly and intellectual disability
Natja Haag, Ene-Choo Tan, Matthias Begemann, Lars Buschmann, Florian Kraft, Petra Holschbach, Angeline H. M. Lai, Maggie Brett, Ganeshwaran H. Mochida, Stephanie DiTroia, Lynn Pais, Jennifer E. Neil, Muna Al-Saffar, Laila Bastaki, Christopher A. Walsh, In
European Journal of Human Genetics.2021; 29(11): 1663. CrossRef
- Exosomes Derived from Bone Marrow Mesenchymal Stem Cells Alleviate Rheumatoid Arthritis Symptoms via Shuttling Proteins
Original Articles
- Molecular Genetic Studies on the Human CYP21A2 Gene (1).
- Byung Kiu Park, Hyang Ok Woo, Han Wook Woo
- J Korean Endocr Soc. 1994;9(3):219-227. Published online November 6, 2019
- 946 View
- 20 Download
-
Abstract
PDF
- Congenital adrenal hyperplasia, especially due to steroid-12-hydroxylase(P450c21) deficiency, is one of the most common autosomal recessive inborn errors at adrenal steroidogenesis in Korean. Molecular genetic analysis has demonstrated that there are two steroid 21-hydroxylase genes, CYP21A1P and CYP21A2. The CYP21A2 gene encodes P450c21, whereas the CYP21A1P gene is a pseudogene. Since there is 98 percent homology between the CYP21A1P and CYP21A2 gene in nucleotide sequences, it has hampered the characterization of molecular defects in the CYP21A2 gene.In this study, efforts have been made to selectively PCR amplify the CYP21A2 gene and test feasibility of DNA microextraction from Guthrie card for prospective use of molecular screening. This study was also aimed at investigating deletion mutations in P450c21 deficient patients, as well as allele frequencies and average heterozygosity of exon 1 A/C polymorphism in Korean newborns. Genomic DNAs were obtained from Guthrie cards of 50 Korean newborns by microextraction method and these DNAs were analyzed by PCR-allele specific oligonucleotide(ASO) hybridization. First part of the CYP21A2 gene has been successfully amplified and digested by restriction enzyme using Taq I or Kpn I, subsequently run on 1.5% agarose gel to confirm its specificity. The anterior 1141 bp PCR product was utilized to examine the frequency and average heterozygosity of exon 1 A/C polymorphism in 100 alleles by ASO dot blot hybridization. Amplified genomic DNAs from four P450c21 deficient patients out of three families were screened by PCR to see if any one has complete deletion of the CYP21A2 gene.The results were as follows;1) The average 1230ng of genomic DNA was obtained form single semi-circled Guthrie card of 1/2 inch diameter by microextraction method, which has been successfully used for DNA analysis.2) The PCR amplified anterior 1141 bp product from the CYP21A2 gene was digested by Kpn I, generating 309 bp, 832 bp fragments, not by Taq I, indicating its specificity.3) The frequencies of exon 1 nucleotide 138 A/C polymorphism in Korean population were 0.81, 0.91 respectively, and average heterozygosity was 0.31.4) None of four P450c21 deficient patients turned out to carry complete deletion of the CYP21A2 gene based on selective PCR amplification of the CYP21A2 gene.In conclusion, dried blood spots from Guthrie card can be sued for DNA analysis because of easy sample collection, bandling, shipment, and DNA extraction feasibility. The selective PCR amplification of the CYP 21A2 gene will pave the way for molecular characterization in P450c21 deficient patients. The exon 1 A/C polymorphism can by efficiently used for molecular diagnosis of P450c21 deficiency in informative families, though it has a drawback of handling radioactive material.
- Endocrine Research
- Functional Identification of Compound Heterozygous Mutations in the CYP17A1 Gene Resulting in Combined 17α-Hydroxylase/17,20-Lyase Deficiency
- Eun Yeong Mo, Ji-young Lee, Su Yeon Kim, Min Ji Kim, Eun Sook Kim, Seungok Lee, Je Ho Han, Sung-dae Moon
- Endocrinol Metab. 2018;33(3):413-422. Published online September 18, 2018
- DOI: https://doi.org/10.3803/EnM.2018.33.3.413
- 3,808 View
- 62 Download
- 2 Web of Science
- 2 Crossref
-
Abstract
PDFPubReader ePub
Background We previously reported a patient with congenital adrenal hyperplasia (CAH) with compound heterozygous mutations in the cytochrome P450 17A1 (
CYP17A1 ) gene. One allele had a p.His373Leu and the other a new p.Glu383fsX36 mutation. The aim of this study was to investigate the functional properties of a new allele present in a compound heterozygote ofCYP17A1 .Methods To understand how p.His373Leu and p.Glu383fsX36 affect P450c17 enzymatic activity, wild type and mutant
CYP17A1 cDNAs were cloned into flag-tagged pcDNA3 vector and introduced into human embryonic kidney cells 293T (HEK293T) cells. Protein expression levels of CYP17A1 were then analyzed. And the activities of 17α-hydroxylase and 17,20-lyase of CYP17A1 were evaluated by measuring the conversion of progesterone to 17α-hydroxyprogesterone and of 17α-hydroxypregnenolone to dehydroepiandrosterone, respectively. In addition a computer model was used to create the three-dimensional structure of the mutant CYP17A1 enzymes.Results Production of the p.His373Leu mutant protein was significantly lower than that of the wild type protein, and the p.Glu383fsX36 protein was hardly produced. Similarly the enzymatic activity derived from the p.His373Leu mutant vector was significantly lower than that obtained from the wild type vector, and little activity was obtained from the p.Glu383fsX36 vector. Three-dimensional modeling of the enzyme showed that p.His373 was located in region important for heme-binding and proper folding. Neither the p.His373Leu nor the p.Glu383fsX36 mutant protein formed a heme-binding structure.
Conclusion Enzyme activity measured in both mutants disappeared completely in both 17α-hydroxylase and 17,20-lyase. This result accounts for the clinical manifestations of the patient with the compound heterozygous
CYP17A1 mutations.-
Citations
Citations to this article as recorded by- A rare case of 17α-hydroxylase/17, 20-lyase deficiency: Clinical and genetic findings and follow-up outcomes
Li-Zhen Dai, Hong Ma, Jian-Fang Ke, Chen-Shi Lin, Yanling Huang, Yuan Tian, Danling Chen
Women's Health.2022; 18: 174550572211225. CrossRef - Novel mutations of the CYP17A1 gene in four Chinese 46,XX cases with partial 17a-hydroxylase/17,20-lyase deficiency
Yanjie Xia, Panlai Shi, Junke Xia, Huijuan Zhang, Lijun Xu, Xiangdong Kong
Steroids.2021; 173: 108873. CrossRef
- A rare case of 17α-hydroxylase/17, 20-lyase deficiency: Clinical and genetic findings and follow-up outcomes
Case Report
- Adrenal gland
- Untreated Congenital Adrenal Hyperplasia with 17-α Hydroxylase/17,20-Lyase Deficiency Presenting as Massive Adrenocortical Tumor
- Su Jin Lee, Je Eun Song, Sena Hwang, Ji-Yeon Lee, Hye-Sun Park, Seunghee Han, Yumie Rhee
- Endocrinol Metab. 2015;30(3):408-413. Published online August 4, 2015
- DOI: https://doi.org/10.3803/EnM.2015.30.3.408
- 4,259 View
- 47 Download
- 3 Web of Science
- 4 Crossref
-
Abstract
PDFPubReader
Congenital adrenal hyperplasia (CAH) with 17α-hydroxylase/17,20-lyase deficiency is usually characterized by hypertension and primary amenorrhea, sexual infantilism in women, and pseudohermaphroditism in men. hypertension, and sexual infantilism in women and pseudohermaphroditism in men. In rare cases, a huge adrenal gland tumor can present as a clinical manifestation in untreated CAH. Adrenal cortical adenoma is an even more rare phenotype in CAH with 17α-hydroxylase/17,20-lyase deficiency. A 36-year-old female presented with hypertension and abdominal pain caused by a huge adrenal mass. Due to mass size and symptoms, left adrenalectomy was performed. After adrenalectomy, blood pressure remained high. Based on hormonal and genetic evaluation, the patient was diagnosed as CAH with 17α-hydroxylase/17,20-lyase deficiency. The possibility of a tumorous change in the adrenal gland due to untreated CAH should be considered. It is important that untreated CAH not be misdiagnosed as primary adrenal tumor as these conditions require different treatments. Adequate suppression of adrenocorticotropic hormone (ACTH) in CAH is also important to treat and to prevent the tumorous changes in the adrenal gland. Herein, we report a case of untreated CAH with 17α-hydroxylase/17,20-lyase deficiency presenting with large adrenal cortical adenoma and discuss the progression of adrenal gland hyperplasia due to inappropriate suppression of ACTH secretion.
-
Citations
Citations to this article as recorded by- Congenital adrenal hyperplasia disorder due to 17 α-hydroxylase deficiency: a case report
Yunling Tian, Lijie Hou, Shulan Xiang, Xuguang Tian, Jinhui Xu
Gynecological Endocrinology.2023;[Epub] CrossRef - Landscape of Adrenal Tumours in Patients with Congenital Adrenal Hyperplasia
Mara Carsote, Ana-Maria Gheorghe, Claudiu Nistor, Alexandra-Ioana Trandafir, Oana-Claudia Sima, Anca-Pati Cucu, Adrian Ciuche, Eugenia Petrova, Adina Ghemigian
Biomedicines.2023; 11(11): 3081. CrossRef - 17α-Hydroxylase/17,20-Lyase Deficiency in 46,XY: Our Experience and Review of Literature
Madhur Maheshwari, Sneha Arya, Anurag Ranjan Lila, Vijaya Sarathi, Rohit Barnabas, Khushnandan Rai, Vishwambhar Vishnu Bhandare, Saba Samad Memon, Manjiri Pramod Karlekar, Virendra Patil, Nalini S Shah, Ambarish Kunwar, Tushar Bandgar
Journal of the Endocrine Society.2022;[Epub] CrossRef - 17α-hydroxylase Deficiency Mimicking Hyperaldosteronism by Aldosterone-producing Adrenal Adenoma
Yun Kyung Cho, Hyeseon Oh, Sun-myoung Kang, Sujong An, Jin-Young Huh, Ji-Hyang Lee, Woo Je Lee
The Korean Journal of Medicine.2016; 91(2): 191. CrossRef
- Congenital adrenal hyperplasia disorder due to 17 α-hydroxylase deficiency: a case report
Original Article
- Six Cases of Congenital Adrenal Hyperplasia That Were Due to 17alpha-hydroxylase/17,20-lyase Deficiency.
- Dong Hoon Shin, Sung Hoon Yu, Young Min Choi, Jung Gu Kim, Sang Wan Kim, Chan Soo Shin, Kyong Soo Park, Seong Yeon Kim
- J Korean Endocr Soc. 2009;24(2):109-115. Published online June 1, 2009
- DOI: https://doi.org/10.3803/jkes.2009.24.2.109
- 1,970 View
- 29 Download
- 2 Crossref
-
Abstract
PDF
- 17alpha-hydroxylase/17,20-lyase deficiency is a rare phenotype of congenital adrenal hyperplasia (CAH), and this is characterized by hyporeninemic hypertension, primary amenorrhea and abnormality of the secondary sexual characteristics (pseudohermaphroditism in men). This type of CAH is usually misdiagnosed at first as mineralocorticoid induced hypertension with primary aldosteronism, but primary amenorrhea with deficient sex hormone is a clue for making the correct diagnosis. The authors experienced 6 cases of 17alpha-hydroxylase/17,20-lyase deficiency in patients who ranged from 15 to 42 years of age. 4 cases were diagnosed according to the investigation of their mineralocorticoid-induced hypertension and 2 cases their primary amenorrhea and sexual infantilism. All of them had hypokalemia, hyporeninemic hypertension and an atrophied uterus and ovaries. In the genotypic male (46 XY), the testicles were atrophied in the abdominal cavity. The levels of cortisol, estrogen and dehydroepiandrosterone sulfate (DHEAS) were low, but the levels of progesterone and 11-deoxycorticosterone were high. Therefore, the diagnosis of 17alpha-hydroxylase/17,20-lyase deficiency should be considered in female patients who present with both sexual infantilism and mineralocorticoid hypertension. We report on these cases with a brief review of the literature.
-
Citations
Citations to this article as recorded by- Functional Identification of Compound Heterozygous Mutations in the CYP17A1 Gene Resulting in Combined 17α-Hydroxylase/17,20-Lyase Deficiency
Eun Yeong Mo, Ji-young Lee, Su Yeon Kim, Min Ji Kim, Eun Sook Kim, Seungok Lee, Je Ho Han, Sung-dae Moon
Endocrinology and Metabolism.2018; 33(3): 413. CrossRef - 17α-hydroxylase Deficiency Mimicking Hyperaldosteronism by Aldosterone-producing Adrenal Adenoma
Yun Kyung Cho, Hyeseon Oh, Sun-myoung Kang, Sujong An, Jin-Young Huh, Ji-Hyang Lee, Woo Je Lee
The Korean Journal of Medicine.2016; 91(2): 191. CrossRef
- Functional Identification of Compound Heterozygous Mutations in the CYP17A1 Gene Resulting in Combined 17α-Hydroxylase/17,20-Lyase Deficiency
Case Reports
- A Case of Congenital Adrenal Hyperplasia Combined with a Testicular Adrenal Rest Tumor and Adrenal Incidentaloma.
- Gyu Rang Cho, Hee Won Chueh, Jung Pyo Kim, Jin A Jung, Jae Ho Yoo, Sung Kook Yoon, Kyu Geun Hwang
- J Korean Endocr Soc. 2007;22(5):365-370. Published online October 1, 2007
- DOI: https://doi.org/10.3803/jkes.2007.22.5.365
- 1,889 View
- 20 Download
- 2 Crossref
-
Abstract
PDF
- The fundamental defect among patients with congenital adrenal hyperplasia (CAH) due to 21-hydroxylse deficiency is the inability to synthesize cortisol and aldosterone adequately. Ineffective cortisol synthesis signals the hypothalamus and pituitary to increase the production of corticotropin releasing hormone and adrenocorticotropic hormone, respectively. Consequently, the adrenal glands become hyperplastic. It is well known that an adrenal adenoma can develop from hyperplastic tissue under increased corticotropin stimulation of the adrenal cortex in patients that are suffering with CAH. The etiologic mechanism of adrenal incidentaloma remains uncertain, but several hypotheses have been suggested. A testicular adrenal rest tumor has been reported to form in association with the excessive secretion of adrenal androgen by inadequate control after adolescence in CAH. We present a case of poorly controlled salt-losing CAH due to 21-hydroxylase deficiency combined with a testicular adrenal rest tumor and adrenal incidentaloma.
-
Citations
Citations to this article as recorded by- A new compound heterozygous mutation in a female with 17α-hydroxylase/17,20-lyase deficiency, slipped capital femoral epiphysis, and adrenal myelolipoma
Fan Yang, Yongting Zhao, Jie Lv, Xia Sheng, Lihong Wang
Gynecological Endocrinology.2019; 35(5): 385. CrossRef - A case of testicular adrenal rest tumor in a male child with congenital adrenal hyperplasia
Joo Hwa Kim, Kyong Ah Yun, Choong Ho Shin, Sei Won Yang
Korean Journal of Pediatrics.2008; 51(9): 1018. CrossRef
- A new compound heterozygous mutation in a female with 17α-hydroxylase/17,20-lyase deficiency, slipped capital femoral epiphysis, and adrenal myelolipoma
- Two Cases of Simple Virilizing Congenital Adrenal Hyperplasia with Compound Heterozygous Mutations of CYP21 Gene.
- Koon Soon Kim, Yun Sun Choi, Youn Sun Bai, So Young Rha, Young Suk Jo, Minho Shong
- J Korean Endocr Soc. 2007;22(4):299-304. Published online August 1, 2007
- DOI: https://doi.org/10.3803/jkes.2007.22.4.299
- 1,877 View
- 34 Download
-
Abstract
PDF
- Steroid 21-hydroxylase deficiency is the most frequent cause of congenital adrenal hyperplasia (CAH), which is an inherited inability to synthesize cortisol. Actually, CAH is caused by mutations in the CYP21 gene encoding the steroid 21-hydroxylase enzyme. In some cases, discordance has been observed between the genotype and the phenotype. We recently experienced two cases of simple virilizing congenital adrenal hyperplasia with compound heterozygous mutations of the CYP21 gene. The patients had primary amenorrhea and showed virilization. We have described these two cases along with a review of the literature.
- A Case of Congenital Adrenal Hyperplasia due to 11beta-Hydroxylase Deficiency.
- Ohk Hyun Ryu, Hye Jin Yoo, Soo Yeon Park, Soon Beom Kwon, Sang Soo Park, Hee Young Kim, Kye Won Lee, Ji A Seo, Jeong Heon Oh, Sin Gon Kim, Nan Hee Kim, Kyung Mook Choi, Sei Hyun Baik, Dong Seop Choi
- J Korean Endocr Soc. 2004;19(1):58-63. Published online February 1, 2004
- 1,222 View
- 35 Download
-
Abstract
PDF
- Congenital adrenal hyperplasia refers to a group of autosomal recessive disorders that is defective in the synthesis of cortisol. The enzymes most often affected are 21-hydroxylase and 11beta hydroxylase. The low levels of cortisol stimulate the pituitary gland to release ACTH. Chronic elevation of the ACTH level causes bilateral adrenal hyperplasia and a secondary increase in androgen formation. We examined a 19 year-old woman presented with clitoral hypertrophy and vaginal spotting. The subjects basal level of serum cortisol was low, but the serum levels of ACTH, 17a-hydroxyprogesterone, deoxy-corticosterone were elevated. The urinary excretions of 17-ketosteroids and 17-hydroxycorticosteroids were also increased. The karyotyping study and transrectal ultrasonography showed normal findings. The patient underwent clitoris reduction surgery and received hydrocortisone. To the best of our knowledge, this is the first case of 11beta-Hydroxylase deficiency in Korea.
- A Case of Primary Amenorrhea due to 17 -Hydroxylase Deficiency.
- Hong Seub Rim, Seon Hwa Lee, Jung Min Hong, Jae Hyun Nam, Hee Back Park, Chul Woo An, Do Min Ki, Sung Kil Lim, Young Duk Song, Hyun Chul Lee, Kap Bum Huh, Inn Soo Kang
- J Korean Endocr Soc. 2001;16(1):130-133. Published online February 1, 2001
- 1,444 View
- 34 Download
-
Abstract
PDF
- 17 -Hydroxylase deficiency is a rare form of congenital adrenal hyperplasia that is characterized by primary amenorrhea, absence of secondary sex characteristics, hypertension, and a hypokalemic alkalosis that has resulted resulting from increased production of deoxycorticosterone and corticosterone by the adrenal. The diagnosis of this enzyme deficiency can be recognized by the increasing serum concentrations of steroid precursors, DOC and corticosterone and the decreasing concentrations of cortisol, and adrenal androgens. We diagnosed this in a 19 year old female who presented with primary amenorrhea. We report this case with a review of the literatures.
Original Articles
- A Study on the Relationship Between Genotype and Phenotype in Korean Patients with Congenital Adrenogenital Syndrome Caused by 21-hydroxylase Deficiency.
- Dong Kyu Jin, Jung Sim Kim, Seung Mi Song, Sung Joon Park, He Zin Hwang, Hwa Young On, Phil Soo Oh, Si Whan Koh, Mee Ryung Uhm, Dong Hwan Lee, Jah Hoon Shin, Heon Seok Han, Hong Sik Kim, Cheol Woo Ko, Han Wook Yoo, Jin Sung Lee, Duk Hee Kim
- J Korean Endocr Soc. 2000;15(2):237-247. Published online January 1, 2001
- 1,050 View
- 28 Download
-
Abstract
PDF
- BACKGROUND
Congenital adrenal hyperplasia (CAH) results from an inherited defect in enzymatic steps required to synthesize cortisol from cholesterol. 21-hydroxylase deficiency accounts for 95% cases of CAH. It appears that the frequency and the type of the responsible mutations differ according to the ethnic background and the type of mutation can predict the clinical outcomes such as salt losing type (SL), simple virilizing type (SV) and non-classic type (NC). METHODS: We have analyzed CYP21 genes in 55 Korean cases (110 chromosomes) of CAH by Southern blotting, PCR-dot hybridization and PCR amplification-created restriction site method. The patients include 43 cases of SL and 12 of SV. None of the NC was found. RESULTS: We found the mutations in 94% (103/110) of the examined chromosomes. A total of 10 types of mutations were discovered. The mutations include aberrant splicing of intron 2 (i2, 35%), CYP21 gene deletion (32%) and I172N (11%) in order. When the relationship between the clinical types and genotypes were correlated, most of the SL patients have either i2 (42%) or CYP21 gene deletion (41%), while SV patients have I172N (33%) or P30L (21%). The parents' mutation was investigated in 20 cases. In 4 families, one of the parents was not the obligatory heterozygote carrier i.e. did not have a mutation. The results suggest the high incidence of de novo mutation. CONCLUSION: We have identified the frequency of mutations of the CYP21 in Korean AGS patients. Our results shows that the clinical type of AGS can be predicted from the genotypes of CYP21. Also the high incidence of de novo mutation of CYP21 confirmed the genetic instability of major histocompatibility III region where the CYP21 is located.
- New Mutation Site in Vasopressin V2 Receptor Gene in a Family with Congenital Nephrogenic Diabetes Incipidus.
- Soon Hee Lee, Chang Hoon Choi, See Hyung Park, Young Sun Choi, Jeong Gook Kim, Seung Woo Ha, Bo Wan Kim
- J Korean Endocr Soc. 2000;15(1):97-106. Published online January 1, 2001
- 1,051 View
- 18 Download
-
Abstract
PDF
- BACKGROUND
Congenital nephrogenic diabetes insipidus (NDI) is a rare inherited disorder, in which two different hereditary forms, X-linked and autosomal recessive traits, have been identified. The X-linked recessive form, mostly (>90%) congenital NDI, has been known to be caused by mutation of the arginine-vasopressin receptor 2 (AVPR2) gene. AVPR2 mutation sites are different in ethnic groups and recently 72 different mutation sites have been reported among AVPR2 gene. This study aimed to analyze AVPR2 gene in selected members in a Korean family with NDI and provided a report of the existence of a new mutation site in AVPR2 gene. METHODS: Three-generation maternal pedigree of the index patient (21-year old male, patient I) and his younger brother (19-year old male, patient II) with NDI was collected. Genomic DNA was obtained from patient I, II, III (index patient's male maternal cousin with NDI), index patient's mother, three maternal aunts, one female maternal cousin and, for control, one healthy male volunteer. Three coding exons of AVPR2 gene were amplified by PCR using 4 pairs of oligonucleotide primers. After direct sequencing of amplified PCR products, the sequence was compared with whole squence of normal AVPR2 gene and identification of a new site of mutation in this gene was done. RESULTS: 1) all three male patients had transversion of G to C at position 1033 of the AVPR2 gene, resulting in a subsequent change of amino acid from glycine to cysteine in codon 201. 2) Two small peaks of G and T, the result of direct sequencing in five female members in this family, would suggest that they are carriers of G to N transversion. CONCLUSION: These results can demonstrate the significant functional correlation of the mutation in AVPR2 gene sequence with clinical NDI, and suggest the clinical utility of direct mutation testing for congenital NDI in family.
 First
First Prev
Prev


