Articles
- Page Path
- HOME > Endocrinol Metab > Volume 30(4); 2015 > Article
-
Case ReportAllgrove (Triple A) Syndrome: A Case Report from the Kashmir Valley
- Raiz Ahmad Misgar, Nazir Ahmad Pala, Mahroosa Ramzan, Arshad Iqbal Wani, Mir Iftikhar Bashir, Bashir Ahmad Laway
-
Endocrinology and Metabolism 2015;30(4):604-606.
DOI: https://doi.org/10.3803/EnM.2015.30.4.604
Published online: December 31, 2015
Department of Endocrinology, Sher-i-Kashmir Institute of Medical Sciences, Srinagar, India.
- Corresponding author: Raiz Ahmad Misgar. Department of Endocrinology, Sher-i-Kashmir Institute of Medical Sciences, Soura, Srinagar, Kashmir 190011, India. Tel: +91-94-1909-0026, Fax: +91-19-4240-3470, drreyaz07@rediffmail.com
• Received: February 7, 2015 • Revised: March 2, 2015 • Accepted: April 13, 2015
Copyright © 2015 Korean Endocrine Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
- Allgrove (Triple A) syndrome is a rare autosomal recessive disorder characterized by cardinal features of adrenal insufficiency due to adrenocorticotropic hormone (ACTH) resistance, achalasia, and alacrimia. It is frequently associated with neurological manifestations like polyneuropathy. Since its first description by Allgrove in 1978, approximately 100 cases have been reported in the literature. Here we report an 18-year-old boy diagnosed as having Allgrove syndrome, with ACTH resistant adrenal insufficiency, achalasia, alacrimia, and severe motor polyneuropathy. Alacrimia was the earliest feature evident at the age of 8 years. He presented with achalasia and adrenal insufficiency at 12 and 18 years respectively and developed neurological symptoms in the form of severe muscle wasting at the age of 15 years. Patients with Allgrove syndrome usually manifest adrenal insufficiency and achalasia during first decade of life. Our patient manifested adrenal insufficiency and achalasia in the second decade and manifested neurological dysfunction before adrenal dysfunction.
- Allgrove for the first time in 1978 described two pairs of siblings with adrenal insufficiency, achalasia cardia and alacrimia [1]. The combination of these three manifestations was known as Allgrove syndrome or Triple A syndrome. The spectrum of disease varies widely and sometimes it is expressed by only two of the three cardinal manifestations. Neurological dysfunction from the involvement of central, peripheral or autonomic nervous systems is often associated with Allgrove syndrome [2]. Adrenal insufficiency and achalasia are usually evident by the end of first decade and neurological dysfunction follows later.
- Triple A syndrome is rare and has been reported from different parts of the world. In the literature it has been described as case reports. The molecular basis of this rare autosomal recessive disorder is the mutated achalasia, adrenocortical insufficiency, alacrimia syndrome (AAAS) gene, located on chromosome 12q13, that codes for ALacrima Achalasia aDrenal Insufficiency Neurologic disorder (ALADIN) protein [3]. Here we present an unusual case of Triple A syndrome in whom neurological manifestations preceded adrenal insufficiency.
INTRODUCTION
- An 18-year-old boy was referred to the endocrine centre at the Sher-i-Kashmir Institute of Medical Sciences, Kashmir for the evaluation of suspected adrenal insufficiency. He was born full term, from a consanguineous marriage should be omitted to a native Kashmiri couple. His younger brother had died at the age of 1 year with possible adrenal insufficiency and his two other younger siblings were healthy (Fig. 1).
- The patient reported generalised weakness, asthenia, fatigue, anorexia and progressive hyperpigmentation of skin for 6 months. Examination revealed a lean build patient (body mass index, 12.64 kg/m2), with hyperpigmentation of tongue, buccal mucosa and skin and a nasal twang to voice. His blood pressure was 100/60 mm Hg with postural drop. Neurological examination revealed wasting of thenar and hypothenar muscles (Fig. 2). The deep tendon reflexes were hyperactive and the sensory examination was unremarkable.
- Baseline investigations revealed normal complete blood counts, serum creatinine and normal electrolytes (serum Na, 142 mmol/L; serum K, 4.1 mmol/L). Basal serum cortisol (8:00 AM) was very low (0.41 µg/dL) and failed to increase after intravenous injection of 250 µg of tetracosactin (30 minutes and 60 values of 0.47 and 0.45 µg/dL, respectively). The plasma adrenocorticotropic hormone (ACTH) levels were markedly elevated (1,262 pg/mL; normal range, 9 to 52 pg/mL). A diagnosis of primary adrenal insufficiency was established. A contrast enhanced computed tomography (CT) of abdomen failed to visualize adrenals indicating atrophic adrenals. The tuberculin test was negative.
- The patient had significant past history. He initially presented to ophthalmologist at the age of 8 years with absence of tears. Schirmer test was suggestive of alacrimia. At that time he had no other complaints and was prescribed artificial tears. Four years later he presented with dysphagia especially to liquids. Barium meal examination revealed persistent narrowing in the distal region of esophagus near gastroesophageal junction. A diagnosis of achalasia cardia was made which was managed with esophagomyotomy. He had again presented at the age of 15 years with severe muscle wasting of thenar, hypothenar muscles and small muscles of feet. Nerve conduction tests revealed axonal peripheral neuropathy.
- On the basis of primary adrenal insufficiency (with normal mineralocorticoid balance), alacrimia, achalasia cardia, and peripheral neuropathy a diagnosis of Allgrove syndrome was established. We have not performed the genetic study about the mutation of AAAS gene. A negative tuberculin test together with absence of adrenal calcification on abdominal CT made tuberculosis unlikely as the basis for adrenal insufficiency in our patient. He was started on oral hydrocortisone tablets 15 mg after breakfast and 10 mg at 6:00 PM. Patient reported a striking improvement on follow up at 4 weeks.
CASE REPORT
- The description of Allgrove syndrome is limited to case reports and thus prevalence is unknown. The molecular basis for this rare autosomal recessive disorder syndrome is the mutated AAAS gene, located on chromosome 12q13, that codes for ALADIN protein. Most of the many reported mutations produce a truncated protein [4].
- The cardinal features of Allgrove syndrome are ACTH resistant adrenal insufficiency, achalasia and alacrimia. The earliest feature is alacrimia as was in our patient. Adrenal insufficiency and achalasia are usually manifested during first decade of life. Achalasia of the cardia occurs in about 75% of cases and in older children/adults it usually manifests as dysphagia especially for liquids. Adrenal insufficiency is also an early manifestation and manifests as severe hypoglycemic or hypotensive attacks during childhood which may lead to sudden death. The death of the younger sibling of our patient at the age of 1 year was possibly due to adrenal insufficiency as he had presented with recurrent vomiting and shock. Adrenal insufficiency in Allgrove syndrome results from a progressive disorder leading to hypofunction of the adrenals at a variable time after birth [5]. The adrenal insufficiency in Allgrove syndrome is a unique form of primary adrenal insufficiency with preservation of mineralocorticoid production. In our patient the documentation of normal electrolytes indicated normal mineralocorticoid production although we have not measured plasma aldosterone levels. The only other cause of primary adrenal insufficiency with preservation of mineralocorticoid production is familial glucocorticoid deficiency. Although most of the patients with Allgrove syndrome have preserved mineralocorticoid production, it may be impaired in 15% of patients [6].
- A neurological syndrome including central, peripheral, and autonomic nervous system impairment is often associated with Allgrove syndrome; neurological manifestations appear at a later age when compared with other manifestations. Distal sensorimotor polyneuropathy is a common manifestation [7]. Our patient presented at the age of 15 years with distal sensorimotor polyneuropathy, 3 years prior to manifesting adrenal insufficiency. Autonomic neuropathy is also frequently associated with Allgrove syndrome but our patient did not manifest any clinical features of autonomic dysfunction. Other neurological manifestations include mental retardation, optic atrophy, amyotrophy, ataxia, dysarthria, hypernasal speech, dementia, parkinsonism, dystonia, and chorea.
- Other features that have been reported in association with Allgrove syndrome include microcephaly, short stature, dysmorphic features (long narrow face, long philtrum, down-turned mouth, thin upper lip, and lack of eyelashes), palmar and plantar hyperkeratosis, osteoporosis, and long QT syndrome [8].
- We emphasize that Allgrove syndrome is a multisystem disease and the cardinal manifestations may appear at any time from infancy to adulthood.
DISCUSSION
-
CONFLICTS OF INTEREST: No potential conflict of interest relevant to this article was reported.
Article information
- 1. Allgrove J, Clayden GS, Grant DB, Macaulay JC. Familial glucocorticoid deficiency with achalasia of the cardia and deficient tear production. Lancet 1978;1:1284–1286. ArticlePubMed
- 2. Clark AJ, Weber A. Adrenocorticotropin insensitivity syndromes. Endocr Rev 1998;19:828–843. ArticlePubMedPDF
- 3. Weber A, Wienker TF, Jung M, Easton D, Dean HJ, Heinrichs C, et al. Linkage of the gene for the triple A syndrome to chromosome 12q13 near the type II keratin gene cluster. Hum Mol Genet 1996;5:2061–2066. ArticlePubMedPDF
- 4. Sandrini F, Farmakidis C, Kirschner LS, Wu SM, Tullio-Pelet A, Lyonnet S, et al. Spectrum of mutations of the AAAS gene in Allgrove syndrome: lack of mutations in six kindreds with isolated resistance to corticotropin. J Clin Endocrinol Metab 2001;86:5433–5437. ArticlePubMed
- 5. Kinjo S, Takemoto M, Miyako K, Kohno H, Tanaka T, Katsumata N. Two cases of Allgrove syndrome with mutations in the AAAS gene. Endocr J 2004;51:473–477. ArticlePubMed
- 6. Grant DB, Barnes ND, Dumic M, Ginalska-Malinowska M, Milla PJ, von Petrykowski W, et al. Neurological and adrenal dysfunction in the adrenal insufficiency/alacrima/achalasia (3A) syndrome. Arch Dis Child 1993;68:779–782. ArticlePubMedPMC
- 7. Goizet C, Catargi B, Tison F, Tullio-Pelet A, Hadj-Rabia S, Pujol F, et al. Progressive bulbospinal amyotrophy in triple A syndrome with AAAS gene mutation. Neurology 2002;58:962–965. ArticlePubMed
- 8. Khong PL, Peh WC, Low LC, Leong LL. Variant of the Triple A syndrome. Australas Radiol 1994;38:222–224. ArticlePubMed
References
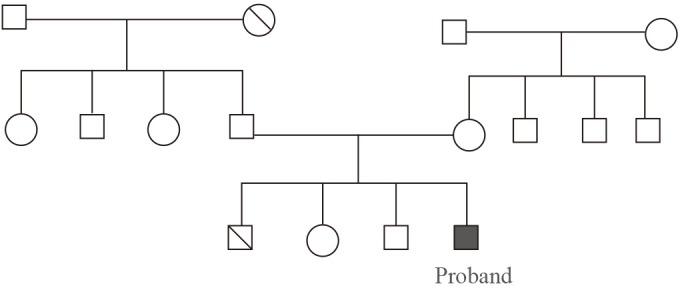
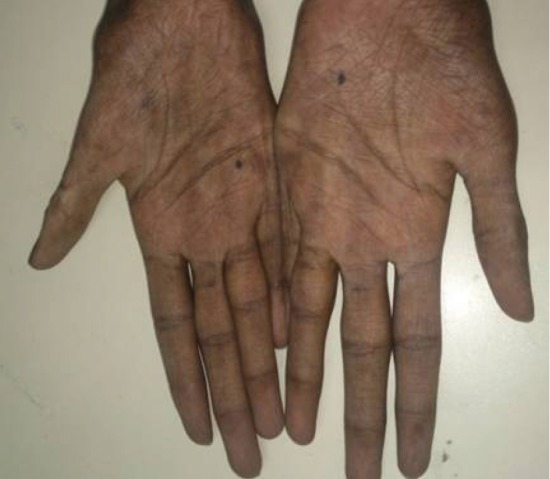
Figure & Data
References
Citations
Citations to this article as recorded by 
- Child Neurology: Allgrove Syndrome
Juhi Gupta, Sayoni Roy Chowdhury, Prashant Jauhari, Kaushik Ragunathan, Biswaroop Chakrabarty, Vandana Jain, Sheffali Gulati
Neurology.2024;[Epub] CrossRef - Don’t forget Allgrove syndrome in adult patients as a bulbar-ALS mimicker
Martina Vigano’, Vittorio Mantero, Paola Basilico, Fiammetta Pirro, Dario Ronchi, Alessio Di Fonzo, Andrea Salmaggi
Neurological Sciences.2023; 44(10): 3703. CrossRef - Triple A (Allgrove) syndrome due to AAAS gene mutation with a rare association of amyotrophy
Satyam Singh Jayant, Rahul Gupta, Kanhaiya Agrawal, Liza Das, Pinaki Dutta, Anil Bhansali
Hormones.2021; 20(1): 197. CrossRef - Allgrove syndrome with amyotrophy
Míriam Carvalho Soares, Otávio Gomes Lins, José Ronaldo Lima de Carvalho, Cláudia Cristina de Sá, Vanessa Van der Linden, Anna Paula Paranhos Miranda Covaleski
Practical Neurology.2021; : practneurol-2021-003192. CrossRef - Triple A syndrome (Allgrove syndrome) – A journey from clinical symptoms to a syndrome
Prakarti Yadav, Deepak Kumar, GopalK Bohra, MahendraK Garg
Journal of Family Medicine and Primary Care.2020; 9(5): 2531. CrossRef - Case report of a familial triple: a syndrome and review of the literature
Federica Gaiani, Pierpacifico Gismondi, Roberta Minelli, Giovanni Casadio, Nicola de’Angelis, Fabiola Fornaroli, Gian Luigi de’Angelis, Marco Manfredi
Medicine.2020; 99(22): e20474. CrossRef - Clinical and genetic characterisation of a series of patients with triple A syndrome
Erdal Kurnaz, Paolo Duminuco, Zehra Aycan, Şenay Savaş-Erdeve, Nursel Muratoğlu Şahin, Melişah Keskin, Elvan Bayramoğlu, Marco Bonomi, Semra Çetinkaya
European Journal of Pediatrics.2018; 177(3): 363. CrossRef - Clinical heterogeneity and molecular profile of triple A syndrome: a study of seven cases
Kanika Singh, Ratna Dua Puri, Pratibha Bhai, Archana Dayal Arya, Garima Chawla, Renu Saxena, Ishwar C. Verma
Journal of Pediatric Endocrinology and Metabolism.2018; 31(7): 799. CrossRef - Clinical and molecular report of c.1331 + 1G > A mutation of the AAAS gene in a Moroccan family with Allgrove syndrome: a case report
H. Berrani, T. Meskini, M. Zerkaoui, H. Merhni, S. Ettair, A. Sefiani, N. Mouane
BMC Pediatrics.2018;[Epub] CrossRef
 PubReader
PubReader Cite
Cite